Синдром меласса. Редкие болезни. Текст научной работы на тему «Эпилепсия при синдроме melas»
Cиндром MELAS относится к митохондриальным болезням (МБ), которые обусловлены генетическими и структурно-биохимическими дефектами митохондрий и сопровождающиеся нарушением тканевого дыхания и, как следствие, системным дефектом энергетического метаболизма, вследствие чего поражаются в различной комбинации наиболее энерго-зависимые ткани и органы-мишени: мозг, скелетные мышцы и миокард, поджелудочная железа, орган зрения, почки, печень. Клинически нарушения в указанных органах могут реализоваться в любом возрасте. При этом гетерогенность симптоматики затрудняет клиническую диагностику этих заболеваний. Необходимость исключения МБ возникает при наличии мультисистемных проявлений, которые не укладываются в обычный патологический процесс. Частоту дисфункции дыхательной цепи оценивают от 1 на 5 - 10 тысяч до 4 - 5 на 100 тысяч новорожденных.
читайте также пост: Митохондриальные болезни (на сайт)
Синдром MELAS (Mitochondrial Encephalomyopathy, Lactic Acidosis and Stroke-like episodes) - это мультисистемное заболевание, которое характеризуется инсультоподобными эпизодами, возникающими в молодом возрасте (до 40 лет), энцефалопатией с судорогами и деменцией, митохондриальной миопатией с феноменом «рваных» красных волокон и лактат-ацидозом (воз- можно повышение уровня молочной кислоты в крови без ацидоза).
В основе синдрома MELAS лежат точковые мутации митохондриальной ДНК (мтДНК). Наследование заболевания осуществляется по материнской линии (следовательно, родственники со стороны матери являются вероятными носителями таких мутаций; чаще у родственников со стороны матери описывают олигосимптоматическую клиническую картину с отдельными симптомами синдрома MELAS; у родственников с бессимптомным течением синдром MELAS идентифицируют только по результатам мышечной биопсии или молекулярного исследования). В настоящее время известно более десяти генов, мутации которых приводят к развитию клинической картины синдрома MELAS. В большинстве случаев к развитию синдрома MELAS приводят мутации в генах, кодирующих функции транспортной РНК.
Обычно заболевание дебютирует в возрасте 6 - 10 лет (возраст начала заболевания - от 3 до 40 лет; раннее начало заболевания является типичным и встречается у 90% пациентов). Характерен низкий рост больных (и непереносимость физических нагрузок). Со стороны внутренних органов может наблюдаться кардиомиопатия, нарушение проводимости сердца, сахарный диабет, нефропатия и нарушение моторики желудочно-кишечного тракта.
Запомните ! Основными клиническими критериями диагноза MELAS являются: [1 ] материнский тип наследования; [2 ] начало до 40 лет; [3 ] нормальное психомоторное развитие до заболевания; [4 ] непереносимость физических нагрузок; [5 ] мигренеподобная головная боль с тошнотой и рвотой; [6 ] инсультоподобные эпизоды; [7 ] энцефалопатия с эпилептическими приступами и/или деменцией (наиболее часто регистрируются миоклонические приступы, однако также отмечаются фокальные сенсорные, моторные и вторично-генерализованные тонико-клонические приступы); [8 ] лактат-ацидоз; [9 ] рваные красные волокна в биоптатах скелетных мышц; [10 ] прогрессирующее течение.
Отличительный клинический признак синдрома MELAS - инсультоподобные эпизоды (ИПЭ), которые являются причиной внезапного развития очаговых неврологических нарушений. Характерная черта ИПЭ - «задняя» локализация очагов поражения в головном мозге. Наиболее часто очаги располагаются в затылочной, теменной и височной долях, реже - в лобной доле, мозжечке или базальных ганглиях; нередко они бывают множественными. Избирательность локализации очаговых изменений обусловливает особенности очаговых неврологических симптомов: гемианопсия, сенсорная афазия, акалькулия, аграфия, оптико-пространственные нарушения, атаксия, изменения сознания ([!!! ] чаще всего очаги локализуются в коре затылочных долей больших полушарий, что приводит к гемианопсии или корковой слепоте). Инсульты могут разрешаться или длительно определяться в виде клинических и/или рентгенологических изменений (что зависит от выраженности метаболических нарушений, обусловленных энергетической недостаточностью нейронов). Часто повторные «инфаркты мозга» развиваются с интервалом в 1 - 3 месяца в симметричных участках. Эти очаги могут быть мелкими или крупными, одиночными или множественными, обычно они несимметричные и их локализация не соответствует зоне кровоснабжения. Кроме того, у пациентов с синдромом MELAS могут наблюдаться кальцификаты в базальных ганглиях (в этих случаях диагностическую помощь может оказать КТ головного мозга). В неврологическом статусе эти морфологические изменения проявляются миоклонусом, атаксией, эпизодами острого психоза или нарушения (дефицита) сознания вплоть до комы ([!!! ] особенность этих острых эпизодов, в т.ч. инсультов, с одной стороны, - быстрый [от нескольких часов до нескольких недель] регресс симптомов, с другой - склонность к рецидивированию); со стороны органов чувств выявляются атрофия зрительных нервов, пигментная ретинопатия и снижение слуха.
Считается, что в генезе ИПЭ имеют значение следующие механизмы: [1 ] метаболические нарушения в мозге с развитием лактат-ацидоза вследствие митохондриальной энергетической недостаточности; [2 ] ишемия мозга, обусловленная митохондриальной ангиопатией на уровне артерий небольшого калибра; [3 ] локальное повышение нейрональной возбудимости вследствие митохондриальной дисфункцией в нейронах, астроцитах или эндотелии капилляров, которая постепенно распространяется по коре головного мозга, сочетается с развитием отека и может привести к ламинарному некрозу в коре мозга.
При поверхностном взгляде инсульт при синдроме MELAS похож на обычный инсульт вследствие тромбоза или эмболии. В действительности инсультоподобные эпизоды при синдроме MELAS являются атипичными: они возникают у молодых людей, часто провоцируются инфекционными заболеваниями, могут протекать в виде мигренеподобной головной боли или судорожных приступов. МРТ-сканирование острых ИПЭ при синдроме MELAS обнаруживает нарушения в виде увеличения сигнала в T2-взвешенном изображении или при FLAIR (инверсия-восстановление с подавлением сигнала от воды) изображении. Повреждения не совпадают с бассейнами крупных мозговых артерий, но в значительной степени затрагивают кору и подлежащее белое вещество с умеренным поражением глубокого белого вещества. Острые повреждения мозга на МР-томограмме при синдроме MELAS могут изменяться, мигрировать или даже исчезать ([!!! ] характерна флуктуация очагов, определяемая при МРТ). Ангиография выявляет отсутствие выраженной сосудистой патологии: помимо нормальных результатов можно обнаружить увеличение калибра артерий, вен или капиллярную гиперемию.
Нейроморфологические исследования мозга при синдроме MELAS показывают наличие мультифокальных некрозов, располагающихся преимущественно в коре больших полушарий головного мозга и подкорковом белом веществе, а также в мозжечке, таламусе и базальных ганглиях. Повреждения напоминают области инфаркта, но, как было указано выше, не совпадают с бассейнами крупных церебральных сосудов. Имеют место также спонгиоформная дегенерация в коре головного мозга, пролиферация капилляров и обеднение нейронами.
Запомните ! Инсультоподобные эпизоды при MELAS имеют следующие особенности: [1 ] молодой возраст (обычно до 40 лет); [2 ] частое наличие провоцирующего фактора (возникают после фебрильной температуры, эпилептического приступа, мигренеподобной головной боли); [3 ] излюбленная локализация - затылочная область; [4 ] очаги, как правило, находятся вне зоны крупных церебральных артерий, чаще располагаясь в коре или глубинных структурах белого вещества головного мозга.
При дифференциальном диагнозе ИПЭ и инфаркта мозга учитываются следующие симптомы:
■ постепенное, на протяжении нескольких дней нарастание очаговых неврологических симптомов (патофизиологической основой такого темпа развития служит постепенное нарастание энергетической недостаточности мозга вследствие нарушения окислительного фосфорилирования в митохондриях);
■ постепенное снижение уровня бодрствования, которое находится в диссонансе с относительно негрубым очаговым неврологическим дефицитом, не сопровождается вторичным стволовым синдромом и, следовательно, не может быть объяснено нарастанием инфаркта мозга и отека (в основе указанных симптомов также лежит расстройство метаболизма, обусловленное нарушением энергетического обеспечения мозга);
■ развитие в остром периоде повторных локальных и генерализованных эпилептических припадков, которые, по данным литературы, встречаются у 2/3 больных с ИПЭ (припадки не связаны с нарушением мозгового кровообращения, так как источником их генерации служит разрядная активность в обоих полушариях головного мозга, а не в структурах, приуроченных к определенному бассейну мозговых артерий; рецидивирующий характер припадков и отсутствие выраженной стойкой очаговой неврологической симптоматики также не характерны для острого инфаркта мозга);
■ полная проходимость артерий мозга по данным дуплексного сканирования брахиоцефальных артерий и церебральной ангиографии, что не типично для ишемического инсульта;
■ особенности нейровизуализационной картины: преимущественно корковая локализация очагов и их «заднее расположение», что характерно для MELAS и объясняется большей уязвимостью нейронов этих областей в связи их большей энергетической потребностью; еще одной нейровизуализационной особенностью служит исчезновение некоторых очагов, в основе которых, по-видимому, лежит отек, а не некроз вещества мозга вследствие метаболических нарушений.
![]()
Cиндром MELAS
на RADIOPAEDIA
.org
Одним из основных проявлений синдрома MELAS также является мышечная слабость (миопатический синдром). Однако неспецифичность этого симптома не позволяет поставить диагноз. Только при возникновении мигрени, судорог и/или инсультоподобных явлений можно диагностировать дебют синдрома MELAS.
Скрининговыми тестами для синдрома MELAS являются нейровизуализация и исследование уровня [повышение] лактата в крови (частично в ликворе) - исследование крови на содержание молочной (лактат) и пировиноградной кислоты (уровень лактата в крови [норма] - венозная кровь - 0,5 - 2,2 ммоль/л, артериальная кровь - 0,5 - 1,6 ммоль/л; соотношение лактат/пируват - 10/1). Диагноз может быть подтвержден исследованием ДНК для определения наиболее частых мутаций. В случае отсутствия часто встречающихся точковых мутаций при синдроме MELAS помочь в диагностике может мышечная биопсия (выявляющая с помощью трехцветного метода Гомори рваные красные волокна [РКВ] - миофибриллы с высоким содержанием мутантного генома и большим числом пролиферирующих измененных митохондрий). Также она (биопсия) позволяет определить наличие биохимических дефектов дыхательной цепи, связанных в основном с ферментами сукцинатдегидро-геназой и цитохромоксидазой.

Лечение синдрома MELAS включает два основных направления. Первый - посиндромная терапия (основное внимание уделяется эпилепсии, сахарному диабету и пр.). Она не отличается от общепринятых подходов к лечению синдромов. Купирование эпилептических приступов необходимо, поскольку метаболический стресс, возникающий при судорогах, может спровоцировать развитие инсультоподобных эпизодов. Широко применяемые в эпилептологии производные вальпроевой кислоты угнетают функции митохондрий, и их применение нежелательно. При невозможности отменить препарат следует одновременно принимать левокарнитин в дозе до 100 мг/кг в сутки. Следует избегать также назначения фенитоина и барбитуратов. Второе направление лечения - патогенетическое, но в настоящее время не существует эффективной патогенетической терапии. Стратегия лечения направлена на улучшение энергетического метаболизма клетки и включает назначение коэнзима Q или идебинона (нобен), препаратов янтарной кислоты, витаминов К1 и К3, никотинамида, рибофлавина, L-карнитина, антиоксидантов (мексидол, милдронат, витамины Е и С), корректоров лактат-ацидоза (димефосфон). [!!!
] Необходимо избегать использования препаратов, угнетающих функцию митохондрий (барбитураты, вальпроаты, статины, глюкокортикоиды).
Подробнее о синдроме MELAS в следующих источниках :
презентация «MELAS-синдром» Кузенкова Л.М., Глоба О.В.; Отделение психоневрологии НИИ педиатрии, Научный Центр Здоровья Детей РАМН, Москва [читать ];
статья «Митохондриальная энцефалопатия с инсультоподобными эпизодами и лактат-ацидозом (синдром MELAS): критерии диагностики, особенности эпилептических приступов и подходы к лечению на примере клинического случая» Ямин М.А., Черникова И.В., Арасланова Л.В., Шевкун П.А.; ГАУ Ростовской области «Областной консультативно-диагностический центр»; кафедра неврологии и нейрохирургии с курсами мануальной терапии и рефлексотерапии ФПК и ППС ФГБОУ ВО «Ростовский государственный медицинский университет» Минздрава России (журнал «Неврология, нейропсихиатрия, психосоматика» №9(4), 2017) [читать ];
статья «Митохондриальные цитопатии: синдромы MELAS и MIDD. Один генетический дефект - разные клинические фенотипы» Муранова А.В., Строков И.А.; ФГБОУ ВО «Первый Московский государственный медицинский университет им. И.М. Сеченова» МЗ РФ, Москва (Неврологический журнал, №1, 2017) [читать ];
статья «Инсультоподобные эпизоды при митохондриальной энцефаломиопатии с лактат-ацидозом» Л.А. Калашникова, Л.А. Добрынина, А.В. Сахарова, Р.П. Чайковская, М.Ф. Мир-Касимов, Р.Н. Коновалов, А.А. Шабалина, М.В. Костырева, В.В. Гнездицкий, С.В. Процкий; Научный центр неврологии РАМН, Москва (журнал «Анналы клинической и экспериментальной неврологии» №3, 2010) [читать ];
статья «Неврологические нарушения при митохондриальной энцефаломиопатии - лактат-ацидозе с инсультоподобными эпизодами (синдроме MELAS)» Д.А. Харламов, А.И. Крапивкин, В.С. Сухоруков, Л.А. Куфтина, О.С. Грознова; Московский НИИ педиатрии и детской хирургии (журнал «Российский вестник перинатологии и педиатрии» №4(2), 2012) [читать ];
статья «Инсультоподобное течение митохондриальной энцефаломиопатии (синдром MELAS)» И.Н. Смирнова, Б.А. Кистенёв, М.В. Кротенкова, З.А. Суслина; Научный центр неврологии РАМН, Москва (журнал «Нервные болезни» №1, 2006) [читать ];
статья «Инсульты при митохондриальных заболеваниях» Н.В. Пизова, Кафедра нервных болезней с курсами нейрохирургии и медицинской генетики ГБОУ ВПО «Ярославская государственная медицинская академия» (журнал «Неврология, нейропсихиатрия, психосоматика» №9(4), 2017) [читать ];
статья «Ишемический инсульт как проявление митохондриальной энцефалопатии у молодого пациента» Мурзалиев А.М., Луценко И. Л., Мусабекова Т.О., Акбалаева Б.A. (журнал «Наука и новые технологии» № 6, 2011) [читать ];
статья «Эпилепсия при синдроме MELAS» Мухин К.Ю., Миронов М.Б., Никифорова Н.В., Михайлова С.В., Чадаев В.А., Алиханов А.А., Рыжков Б.Н., Петрухин А.С.; ГОУ ВПО РГМУ Росздрава; Российская детская клиническая больница (Русский журнал детской неврологии» №3, 2009) [читать ];
статья «Алгоритм диагностики митохондриальных энцефаломиопатий» С.Н. Иллариошкин, НИИ неврологии РАМН (журнал «Нервные болезни» №3, 2007) [читать ]
© Laesus De Liro
Синдром МЕЛАС — это митохондриальное заболевание, характеризующееся поражением мышц и ЦНС.
MELAS (англ. Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes - «митохондриальная энцефаломиопатия, лактатацидоз, инсультоподобные эпизоды») - прогрессирующее нейродегенеративное заболевание, характеризующееся проявлениями, перечисленными в названии, и сопровождается полиморфной симптоматикой - инсульта, диабетом, судорогами, снижением слуха, сердечными заболеваниями, низким ростом, эндокринопатиями, непереносимостью физических нагрузок и нейропсихиатрическими отклонениями.
История
.
Синдром MELAS был впервые описан в 1984 году Павлакисом и коллегами; десять лет спустя Павлакис и Мицио Хирано опубликовали обзор 110 случаев заболевания.
Тип наследования:
материнский
Эпидемиология :
Точная частота заболевания не известна. В литературе имеются единичные данные о частоте заболевания. На севере Финляндии частота мутации A3243G, составляет 16.3:100 000.
Патогенез :
Мутации митохондральных ДНК, контролирующих дыхательную цепь митохондрий, сопровождаются нарушением процессов окислительного фосфорилирования — важнейшего источника энергии для метаболических процессов в клетке.
Клинические проявления
В возрасте до 40 лет пациенты с МЕЛАС поступают с клиникой транзиторной ишемической атаки, а также с эпилепсией, неоднократной рвоты, головной болью, мышечную слабость. У данных пациентов нередко клинически выявляют деменцию.
Молодой возраст и отсутствие факторов риска, характерных для инсульта, помогает задуматься о МЕЛАС.
Лабораторные данные
Лактат ацидоз — увеличения уровня лактата и пирувата.
Данные визуализации
Изменения головного мозга схожи с изменениями при инсульте.
Отличия от инсульта
1) области поражения не совпадают с границами артериальных сосудистых бассейнов.
2) при повторных приступах очаги визуализируются в другой локализации.
+ клинические данные (молодой возраст, отсутствие факторов риска инсульта).
КТ
Множественные гиподенсивные области не соответствующие сосудистому бассейну.
Кальцификация базальных ганглий (наиболее чаще у пожилых пациентов).
Атрофия возникает на фоне регресса и клинического улучшения.
МРТ
Острый инфаркт
Для дифференциации с инсультом используют ADC и DWI (при инсультах ограничение диффузии (цитотоксический отек), а при МЕЛАС диффузия ограничена незначительно, либо без изменений (вазогенный отек).
Вовлечение в патологический процесс субкортикального белого вещества головного мозга.
Ухудшение визуализации четкости контуров извилин и повышение сигнала от них на Т2-взвешенных изображения.
Хронический инфаркт
Изменения могут быть симметричными и ассиметричными.
Фокальная атрофия возникает на фоне регресса и клинического улучшения.
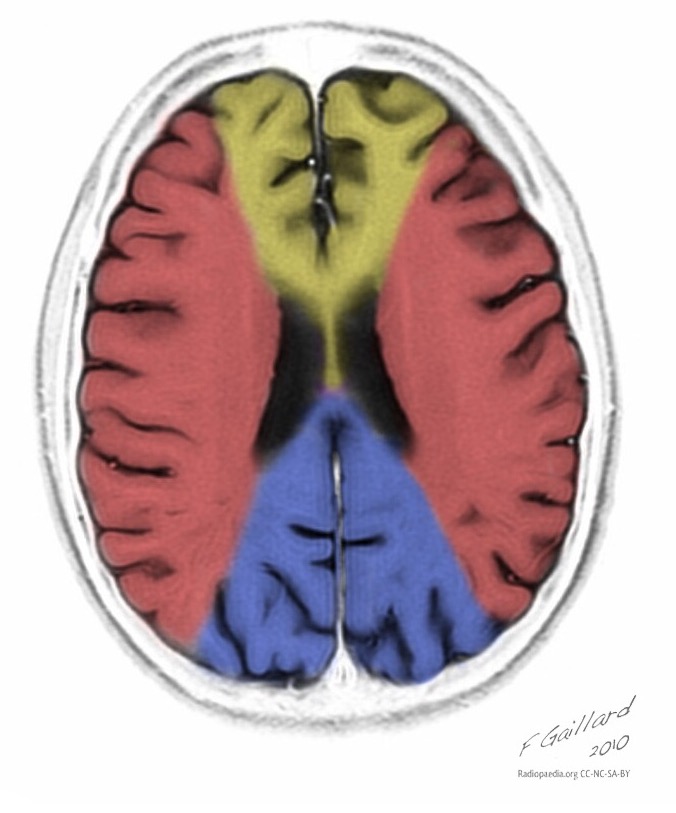
Теменная, затылочная и височная доля головного мозга наиболее чаще поражаются.
МР-спектроскопия
Повышение уровня лактата.

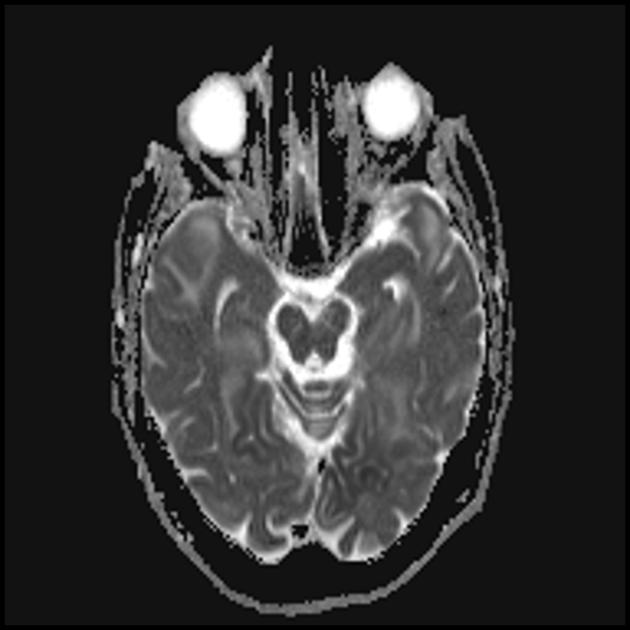

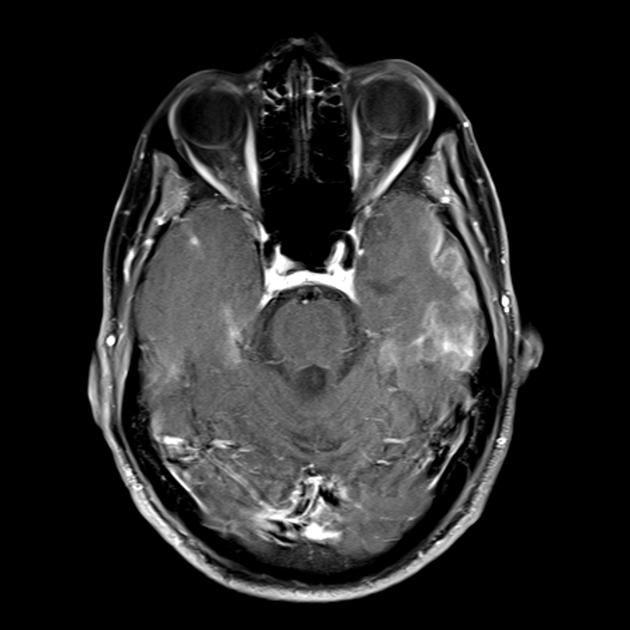
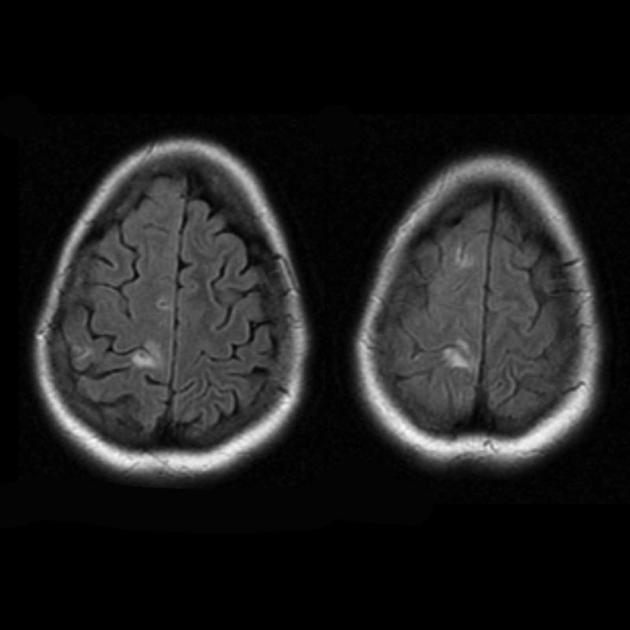

Синдром MELAS (mitochondrial encephalomyopathy, lactic acidosis and stroke) — митохондриальная энцефаломиопатия, лактат-ацидоз и инсульт — выделен сравнительно недавно (в 1984 г.). Заболевание связано с точковой мутацией митохондриальной ДНК, которая в 90% локализуется в гене, кодирующем синтез транспортной РНК лейцина, что препятствует его включению в белки дыхательной цепи. Как и при всех митохондриальных заболеваниях, диагностика синдрома MELAS представляет трудности, обусловленные значительным разнообразием клинической картины.
Основные клинические проявления синдрома MELAS: непереносимость физических нагрузок; рецидивирующие инсультоподобные состояния. Термин «инсультоподобные», вероятно, обусловлен тем, что в большинстве описываемых случаев ведущим клиническим проявлением является головная боль с рвотой, судорогами, часто с нарушением сознания, длительностью от нескольких часов до нескольких дней.
Во время этих атак у ряда больных нередко развиваются неврологические нарушения в виде гемианопсии, гемипареза, редко в виде афазии. При проведении КТ у 60% таких пациентов выявляются очаги пониженной плотности, полиморфные судорожные приступы; при биохимическом исследовании выявляется лактат-ацидоз; начало заболевания — в 5-6-летнем возрасте; течение болезни носит прогрессирующий характер.
Приводим собственное наблюдение MELAS синдрома.
Больной А., 6 лет, поступил в клинику с жалобами на остро развившуюся слабость в правых конечностях, которая удерживалась в течение нескольких часов, головные боли. Родился от первой беременности. Беременность и роды протекали без осложнений. Раннее психомоторное развитие ребенка соответствовало возрасту. С 1 года до 2 лет отмечались аффективно-респираторные пароксизмы. С 3 лет частые ацетонемические состояния, развивающиеся на высоте головной боли.
Во время осмотра в клинике: двигательная расторможенность, неусидчивость, быстрая утомляемость при нагрузках (как умственных, так и физических). В неврологическом статусе: черепная иннервация без особенностей, мышечная гипотония, асимметрия сухожильных рефлексов (более живые справа). Парезов нет. Атаксии нет.
При первичном проведении МРТ головного мозга в левой затылочной доле обнаружена обширная зона гиперинтенсивного сигнала в режиме Т2 и гипоинтенсивность в режиме Т1 с четкими контурами, срединные структуры не смещены. Данная радиологическая картина была расценена как явление ишемического инсульта.
В течение последующего года у ребенка трижды отмечались парциальные эпилептические приступы, повторялись эпизоды приступообразной головной боли с рвотой. Получал противосудорожную терапию. Спустя год после первого лечения в клинике мальчик поступил повторно в связи с развившимся приступом генерализованных тонико-клонических судорог, который в течение часа повторился дважды.
Наросла вялость, слабость, появилась приступообразная головная боль с однократной рвотой. Повторно проведена МРТ головного мозга — в режимах Т1 иТ2 визуализировались участки измененного сигнала в теменно-затылочных отделах с обеих сторон (причем очаг слева имел меньшие размеры по сравнению с таковым при предыдущем МРТ-исследовании). Таким образом, за прошедшие после первого инсульта несколько месяцев мальчик перенес по крайней мере еще три острых нарушения мозгового кровообращения.
Метаболические нарушения заключались в значительном увеличении уровня лактата в крови до 4,2 ммоль/л (норма до 1,7 ммоль/л) и пирувата.
Комплексный анализ результатов обследования и клиники позволил установить конкретную нозологическую форму митохондриальной энцефаломиопатии — синдром MELAS, который в клинике был установлен впервые.
В последующем ребенок получал противосудорожную терапию (топамакс 5 мг/кг/сут.), лечение, направленное на стимуляцию тканевого дыхания с использованием коэнзима Q-10, янтарной кислоты, цитомака, а также актовегин и контрикал.
Приведенный случай рецидивирующих метаболических инфарктов мозга у ребенка, обусловленных синдромом MELAS, не является единственным случаем в клинике, и банк данных о подобных больных накапливается.
Материалы рассчитаны на врачей-неврологов, терапевтов, общей практики.
Сергей Лихачёв, заведующий, доктор мед. наук, профессор;
Инесса Плешко, ведущий научный сотрудник, кандидат мед. наук, неврологический отдел РНПЦ неврологии и нейрохирургии.
Церебральная аутосомно-доминантная артериопатия с субкортикальными инфарктами и лейкоэнцефалопатией (CADASIL) -прогрессирующее аутосомно-доминантное заболевание, клинические проявления которого включают повторные подкорковые ишемические инсульты, мигрень, субкортикальную деменцию и аффективные нарушения. Распространенность в настоящее время - 1 случай
на 100 000 населения.
В РНПЦ неврологии и нейрохирургии наблюдаются 7 пациентов (в т. ч. 4 женщины) с CADASIL; возраст - от 32 до 68 лет. Их обследовали неврологическим, молекулярно-генетическим методами. Имела место характерная симптоматика; в анамнезе - мигрень, рецидивирующие лакунарные инсульты и аффективные нарушения. МРТ головного мозга позволила обнаружить свойственные CADASIL субкортикальные инфаркты и лейкоэнцефалопатию.
У 2 человек в результате молекулярно-генетической диагностики выявлена гетерозиготная мутация в гене Notch3 на 19-й хромосоме, чем и вызывается CADASIL. Гены Notch кодируют трансмембранные рецепторы, участвующие в онтогенезе клетки. При CADASIL в большинстве случаев определяют миссенс-мутации, из-за которых изменяются структуры трансмембранного белка и нарушаются его функции.
Патогенез CADASIL окончательно не ясен. Считается, что основной фактор - артериопатии с прогрессирующей окклюзией мелких перфорирующих сосудов белого вещества головного мозга (приводит к хронической гипоперфузии). При этом обнаруживают характерные гранулярные осмиофильные включения, вызывающие пролиферацию компонентов базальной мембраны, утолщение средней оболочки и механическое сдавление мелких артерий. В итоге повреждается гематоэнцефалический барьер - развивается отек.
Дополнительный патологический фактор - активация астроцитов вблизи сосудистой стенки. Они высвобождают эндотелии-1, провоцируя вазоконстрикцию и нарушение кровотока.
Состав гранулярных осмиофильных включений неизвестен. Предполагается, что белок Notch3 является одним из их компонентов. При биопсии кожи пациентов с мутацией в гене Notch3 осмиофильные гранулы и дегенерация гладких мышечных клеток могут определяться еще до 20-летнего возраста.
Клиническая диагностика CADASIL:
- отягощенный семейный анамнез;
- развитие первых cимптомов заболевания до 50 лет;
- присутствие двух из сле-дующих симптомов - мигрень, повторные инсульты, нарушения настроения, субкортикальная деменция.
Следует исключить сосудистые факторы риска, этиологически связанные с неврологическими симптомами. МРТ показывает поражение белого вещества полушарий головного мозга и отсутствие кортикальных инфарктов.
Достоверный диагноз «CADASIL» подтверждается положительным результатом молекулярно-генетической диагностики или обнаружением артериопатии с характерными гранулярными осмиофильными включениями при биопсии кожи или мышцы.
Самые частые симптомы CADASIL - преходящие ишемические атаки и ишемические инсульты, наблюдаемые почти у 85% пациентов.
Характеризуются рецидивирующим течением, проявляются классическими синдромами лакунарных инсультов и полной клинической ремиссией через несколько дней или недель.
Вторые по частоте - когнитивные нарушения (отмечаются у 60% пациентов). Могут начинаться в 35 лет, иногда еще до ишемических эпизодов. Приблизительно в 75% случаев при CADASIL развивается деменция. Первый симптом - обычно мигрень; часто возникает до 20 лет и, как правило, предшествует инсультам.
Данные о вовлечении сердца в патологический процесс при CADASIL противоречивые. L. Oberstein et al. (2003 г.) обнаружили, что 25% пациентов с диагностированным CADASIL имели острый инфаркт миокарда в анамнезе или патологию Q-волны на электрокардиограмме. В другом исследовании Cumurciuc et al. (2006 г.) не выявили положительного кардиологического анамнеза у 23 человек с мутацией в гене Notch3.
Клинические проявления CADASIL и микроангиопатии головного мозга иной этиологии схожи - требуется дифференциальная диагностика.
Чтобы своевременно определить CADASIL у пациентов и членов их семей, надо прибегать к молекулярно-генетическим методам и/или гистологическим исследованиям.
Синдром MELAS
Митохондриальная энцефаломиопатия с лактат-ацидозом и инсультоподобными эпизодами (MELAS) - редкое наследственное заболевание, обусловленное патологией митохондриального генома, нарушением энергетического метаболизма и функционирования наиболее энергозависимых органов и тканей (ЦНС, сердечная и скелетные мышцы, глаза, почки, печень, костный мозг, эндокринная система). Широкая вариабельность клинических проявлений MELAS-синдрома и редкая встречаемость предопределяют трудности при диагностике для практического врача.
В РНПЦ неврологии и нейрохирургии наблюдаются 3 пациента (46-летняя женщина и ее сыновья - 24 и 23 лет) с диагностированным синдромом MELAS. Они прошли клинико-неврологическое обследование, молекулярно-генетическую диагностику, МРТ головного мозга.
У всех низкорослость; в анамнезе - симптомы митохондриальной патологии: нейросенсорная тугоухость, мигренеподобные головные боли, плохая переносимость физических нагрузок. Дебют заболевания - генерализованные судорожные приступы. У 2 пациентов первые симптомы проявились до 20 лет; имели место эпилептические припадки, следующие один за другим, эпизоды нарушения зрения с наличием очагов при нейровизуализации в затылочных и височных областях, повышение уровня лактата в крови и цереброспинальной жидкости. У 1 человека выявлено умеренное снижение когнитивных функций; по данным УЗИ сердца - гипертрофическая кардиомиопатия; сахарный диабет.
При молекулярно-генетическом исследовании обнаружены типичные для MELAS мультисистемность поражения, широкая вариабельность и различная степень выраженности клинических проявлений, соответствующие количеству мутантных копий А3243G в тРНКLeu(UUR) гене.
Для MELAS характерны материнский тип наследования, наличие спорадических случаев при возникновении мутации de novo; накопление в клетках - как нормальных, так и мутантных типов - митохондриальной ДНК (гетероплазмия) и случайное распределение при делении между дочерними клетками (митотическая сегрегация). На генетическом уровне причина MELAS-синдрома - гетероплазмическая перестановка 3243A>G в тРНКLeu(UUR) гене (обнаруживается 80% случаев).
Патогенез заболевания пока не изучен. Существуют 2 основные теории - «митохондриальной ангиопатии» и «митохондриальной цитопатии». Известно, что инсультоподобное поражение не соответствует сосудистым зонам и распространяется на окружающие области вследствие сопутствующего вазогенного отека, вызванного длительной эпилептической активностью. Как предполагают, инсультоподобные эпизоды обусловлены нейронной гипервозбудимостью в ограниченном участке мозга. Она возникает из-за митохондриальной дисфункции в эндотелиальных клетках капилляров, или в нейронах, или в астроцитах; деполяризует смежные нейроны, приводя к распространению эпилептической активности.
Кроме того, в промежутках между инсультоподобными эпизодами, по данным однофотонно-эмиссионной компьютерной томографии (SPECT), у пациентов с MELAS наблюдается гипоперфузия коры задней части поясной извилины, что свидетельствует о расстройстве мозговой гемодинамики.
Нарушение окислительного фосфорилирования, разрыв митохондриальной дыхательной цепи способствуют тому, что преобладают катаболический метаболизм и изменения от цикла Кребса к анаэробному гликозу с накоплением лактата. Высокий уровень последнего в ЦНС обычно коррелирует с периодами неврологической симптоматики.
Основные клинические признаки MELAS - инсультоподобные эпизоды, лактат-ацидоз, наличие в мышечных биоптатах «рваных красных волокон». Дополнительными проявлениями могут быть деменция, психозы, эпилептические пароксизмы, мигренеподобные головные боли, атаксия, миопатия, кальцификация базальных ганглиев по данным нейровизуализации, оптическая атрофия, ретинопатия, глухота, диабет, интестинальная псевдообструкция, кардиомиопатия.
Ранний возраст дебюта MELAS - от 5 до 20 лет, однако есть наблюдения и позднего начала - на 5–6-й декадах жизни. Известны случаи, когда синдром запускался после кардиологических нарушений.
Мультисистемность поражения при MELAS осложняет клиническую диагностику.
Наследственный характер заболевания обязывает провести молекулярно-генетические исследования, чтобы поставить точный диагноз
и выявить других пациентов - из числа родственников больного.
Материалы рассчитаны на врачей-неврологов, терапевтов, общей практики.
 (1 оценок, в среднем: 5,00 из 5)
(1 оценок, в среднем: 5,00 из 5)