Синдром на меласа. Редки болести. Текст на научната работа по темата "Епилепсия при мелас синдром"
Синдромът MELAS се отнася до митохондриални заболявания (MD), които се причиняват от генетични и структурно-биохимични дефекти на митохондриите и са придружени от нарушено тъканно дишане и, като следствие, системен дефект в енергийния метаболизъм, в резултат на което най-много енергия -зависимите тъкани и таргетните органи се засягат в различни комбинации: мозък, скелетни мускули и миокард, панкреас, орган на зрението, бъбреци, черен дроб. Клиничните нарушения в тези органи могат да възникнат във всяка възраст. В същото време хетерогенността на симптомите усложнява клиничната диагноза на тези заболявания. Необходимостта от изключване на MB възниква при наличие на мултисистемни прояви, които не се вписват в обичайния патологичен процес. Честотата на дисфункцията на дихателната верига се оценява от 1 на 5 - 10 хиляди до 4 - 5 на 100 хиляди новородени.
прочетете и поста: Митохондриални заболявания(към уебсайта)
Синдромът MELAS (митохондриална енцефаломиопатия, лактатна ацидоза и инсулт-подобни епизоди) е мултисистемно заболяване, характеризиращо се с инсулт-подобни епизоди, възникващи в млада възраст (преди 40-годишна възраст), енцефалопатия с припадъци и деменция, митохондриална миопатия с феномена „ накъсани” червени влакна и лактатна ацидоза (възможно е да се повиши нивото на млечна киселина в кръвта без ацидоза).
Синдромът на MELAS се причинява от точкови мутации в митохондриалната ДНК (mtDNA). Заболяването се унаследява по майчина линия (следователно роднините по майчина линия вероятно са носители на такива мутации; по-често роднините по майчина линия описват олигосимптоматична клинична картина с отделни симптоми на синдрома на MELAS; при асимптоматични роднини синдромът на MELAS е идентифицирани само чрез резултатите от мускулна биопсия или молекулярно изследване). Понастоящем са известни повече от десет гена, чиито мутации водят до развитието на клиничната картина на синдрома MELAS. В повечето случаи развитието на синдрома MELAS се причинява от мутации в гени, кодиращи функциите на трансферната РНК.
Обикновено заболяването дебютира на възраст между 6 и 10 години (възрастта на началото е от 3 до 40 години; характерно е ранното начало на заболяването и се среща при 90% от пациентите). Пациентите се характеризират с нисък ръст (и непоносимост към упражнения). От страна на вътрешните органи могат да се наблюдават кардиомиопатия, нарушения на сърдечната проводимост, захарен диабет, нефропатия и нарушена подвижност на стомашно-чревния тракт.
Помня! Основните клинични критерии за диагностициране на MELAS са: [ 1 ] тип наследство по майчина линия; [ 2 ] начало преди 40-годишна възраст; [ 3 ] нормално психомоторно развитие преди заболяването; [ 4 ] упражнява непоносимост; [ 5 ] мигреноподобно главоболие с гадене и повръщане; [ 6 ] инсулт-подобни епизоди; [ 7 ] енцефалопатия с епилептични припадъци и/или деменция (най-често се регистрират миоклонични припадъци, но се отбелязват и фокални сензорни, моторни и вторично генерализирани тонично-клонични припадъци); [ 8 ] лактатна ацидоза; [ 9 ] накъсани червени влакна в биопсии на скелетни мускули; [ 10 ] прогресивен курс.
Отличителната клинична характеристика на синдрома MELAS са инсулт-подобни епизоди (IPE), които причиняват внезапно развитие на фокални неврологични разстройства. Характерна особеност на IPE е "задната" локализация на лезиите в мозъка. Най-често лезиите се локализират в тилния, париеталния и темпоралния лоб, по-рядко във фронталния лоб, малкия мозък или базалните ганглии; често те са множество. Селективността на локализацията на фокалните промени определя характеристиките на фокалните неврологични симптоми: хемианопсия, сензорна афазия, акалкулия, аграфия, оптико-пространствени нарушения, атаксия, промени в съзнанието ([ !!! ] най-често лезиите са локализирани в кората на тилните дялове на мозъчните хемисфери, което води до хемианопсия или кортикална слепота). Инсултите могат да отзвучат или да бъдат дългосрочно определени под формата на клинични и/или радиологични промени (което зависи от тежестта на метаболитните нарушения, причинени от енергиен дефицит на невроните). Често повтарящи се "мозъчни инфаркти" се развиват на интервали от 1 до 3 месеца в симетрични зони. Тези лезии могат да бъдат малки или големи, единични или множествени, обикновено са асиметрични и локализацията им не съответства на областта на кръвоснабдяването. В допълнение, пациентите със синдром на MELAS може да имат калцификати в базалните ганглии (в тези случаи компютърната томография на мозъка може да бъде полезна). В неврологичния статус тези морфологични промени се проявяват с миоклонус, атаксия, епизоди на остра психоза или нарушение (дефицит) на съзнанието до кома ([ !!! ] характеристика на тези остри епизоди, вкл. инсулти, от една страна, бърза [от няколко часа до няколко седмици] регресия на симптомите, от друга, тенденция към рецидив); от сетивните органи се откриват атрофия на зрителните нерви, пигментна ретинопатия и загуба на слуха.
Смята се, че следните механизми са важни за генезиса на IPE: [ 1 ] метаболитни нарушения в мозъка с развитие на лактатна ацидоза поради митохондриален енергиен дефицит; [ 2 ] церебрална исхемия, причинена от митохондриална ангиопатия на ниво малки артерии; [ 3 ] локално повишаване на невроналната възбудимост поради митохондриална дисфункция в неврони, астроцити или капилярен ендотел, което постепенно се разпространява в церебралния кортекс, се комбинира с развитието на оток и може да доведе до ламинарна некроза в мозъчната кора.
На повърхностен поглед, инсулт със синдрома на MELAS е подобен на нормален инсулт, дължащ се на тромбоза или емболия. Всъщност подобните на инсулт епизоди при синдрома на MELAS са нетипични: те се срещат при млади хора, често се провокират от инфекциозни заболявания и могат да се появят под формата на мигреноподобно главоболие или гърчове. MRI сканиране на остър IPE при синдром на MELAS разкрива аномалии като повишен сигнал на T2-претеглени или FLAIR (водно-атенюирано възстановяване при инверсия) изображения. Лезиите не съвпадат с териториите на главните церебрални артерии, но до голяма степен включват кората и подлежащото бяло вещество, с умерено засягане на дълбокото бяло вещество. Острите мозъчни лезии на ЯМР при синдром на MELAS могат да се променят, мигрират или дори да изчезнат ([ !!! ] характеризиращ се с флуктуация на огнищата, определени с ЯМР). Ангиографията разкрива липсата на значителна съдова патология: в допълнение към нормалните резултати може да се открие увеличение на калибъра на артериите, вените или капилярна хиперемия.
Невроморфологичните изследвания на мозъка при синдром на MELAS показват наличието на мултифокална некроза, локализирана главно в мозъчната кора и субкортикалното бяло вещество, както и в малкия мозък, таламуса и базалните ганглии. Лезиите приличат на зони на инфаркт, но, както бе споменато по-горе, не съвпадат с басейните на големите церебрални съдове. Има също спонгиформна дегенерация в мозъчната кора, пролиферация на капиляри и изчерпване на невроните.
Помня! Подобните на инсулт епизоди при MELAS имат следните характеристики: [ 1 ] млада възраст (обикновено до 40 години); [ 2 ] често наличие на провокиращ фактор (възниква след фебрилна температура, епилептичен пристъп, мигреноподобно главоболие); [ 3 ] любима локализация - тилна област; [ 4 ] лезиите, като правило, се намират извън областта на големите церебрални артерии, най-често разположени в кората или дълбоките структури на бялото вещество на мозъка.
При диференциална диагноза на IPE и мозъчен инфаркт се вземат предвид следните симптоми:
■ постепенно, в продължение на няколко дни, увеличаване на фокалните неврологични симптоми (патофизиологичната основа за този темп на развитие е постепенното увеличаване на енергийния дефицит на мозъка поради нарушено окислително фосфорилиране в митохондриите);
■ постепенно намаляване на нивото на будност, което е в дисонанс с относително лек фокален неврологичен дефицит, не е придружено от вторичен синдром на мозъчния ствол и следователно не може да се обясни с увеличаване на мозъчния инфаркт и оток (тези симптоми също са въз основа на метаболитно разстройство, причинено от нарушение на енергийното снабдяване на мозъка);
■ развитие в острия период на повтарящи се локални и генерализирани епилептични припадъци, които според литературата се срещат при 2/3 от пациентите с IPE (припадъците не са свързани с мозъчно-съдов инцидент, тъй като източникът на тяхното генериране е активността на освобождаване от отговорност в двете полукълба на мозъка, а не в структури, ограничени до конкретен басейн на церебрални артерии; повтарящият се характер на гърчовете и липсата на изразени персистиращи фокални неврологични симптоми също не са характерни за острия мозъчен инфаркт);
■ пълна проходимост на мозъчните артерии според дуплексно сканиране на брахиоцефални артерии и церебрална ангиография, което не е типично за исхемичния инсулт;
■ характеристики на невроизобразителната картина: предимно кортикална локализация на огнищата и тяхното „задно разположение“, което е характерно за MELAS и се обяснява с по-голямата уязвимост на невроните в тези области поради по-голямата им енергийна нужда; Друга характеристика на невроизображението е изчезването на някои огнища, което очевидно се основава на оток, а не на некроза на мозъчното вещество поради метаболитни нарушения.
![]()
MELAS синдромНа РАДИОПЕДИЯ.org
Една от основните прояви на синдрома MELAS също е мускулна слабост (миопатичен синдром). Въпреки това, неспецифичността на този симптом не позволява диагноза. Само когато възникнат мигрена, гърчове и/или подобни на инсулт събития, може да се диагностицира началото на синдрома на MELAS.
Скрининговите тестове за синдрома на MELAS са невроизобразяване и изследване на нивото [увеличаване] на лактат в кръвта (частично в цереброспиналната течност) - кръвен тест за млечна (лактат) и пирогроздена киселина (ниво на лактат в кръвта [нормално] - венозна кръв - 0,5 - 2,2 mmol/l, артериална кръв - 0,5 - 1,6 mmol/l, съотношение лактат/пируват - 10/1). Диагнозата може да бъде потвърдена чрез ДНК изследване за определяне на най-честите мутации. При липса на общи точкови мутации при синдрома на MELAS, мускулна биопсия (използвайки трицветния метод на Gomori за откриване на накъсани червени влакна [RRFs] - миофибрили с високо съдържание на мутантен геном и голям брой пролифериращи променени митохондрии) може да помогне в диагнозата. Тя (биопсия) също така позволява да се определи наличието на биохимични дефекти в дихателната верига, свързани главно с ензимите сукцинат дехидрогеназа и цитохром оксидаза.

Лечението на синдрома MELAS включва две основни области. Първата е синдромна терапия (фокусът е върху епилепсия, диабет и др.). Не се различава от общоприетите подходи за лечение на синдроми. Спирането на епилептичните припадъци е необходимо, тъй като метаболитният стрес, който възниква по време на припадъците, може да предизвика развитието на епизоди, подобни на инсулт. Производните на валпроевата киселина, широко използвани в епилептологията, инхибират митохондриалните функции и употребата им е нежелателна. Ако е невъзможно да спрете лекарството, трябва едновременно да приемате левокарнитин в доза до 100 mg / kg на ден. Фенитоинът и барбитуратите също трябва да се избягват. Второто направление на лечение е патогенетично, но в момента няма ефективна патогенетична терапия. Лечебната стратегия е насочена към подобряване на енергийния метаболизъм на клетката и включва прилагане на коензим Q или идебинон (Noben), препарати от янтарна киселина, витамини K1 и K3, никотинамид, рибофлавин, L-карнитин, антиоксиданти (мексидол, милдронат, вит. Е и С), лактатни коректори -ацидоза (димефосфон). [ !!!
] Необходимо е да се избягва употребата на лекарства, които инхибират митохондриалната функция (барбитурати, валпроати, статини, глюкокортикоиди).
Прочетете повече за синдрома на MELAS в следните източници:
презентация „Синдром MELAS” Кузенкова Л.М., Глоба О.В.; Катедра по психоневрология, Изследователски институт по педиатрия, Научен център за детско здраве, Руската академия на медицинските науки, Москва [прочетете];
статия „Митохондриална енцефалопатия с инсулт-подобни епизоди и лактатна ацидоза (MELAS синдром): диагностични критерии, характеристики на епилептичните припадъци и подходи за лечение на примера на клиничен случай” Yamin M.A., Chernikova I.V., Araslanova L.V., Shevkun P.A.; Държавна автономна институция на Ростовска област „Регионален консултативен и диагностичен център“; Катедра по неврология и неврохирургия с курсове по мануална терапия и рефлексология на Факултета по образование и обучение на Федералната държавна бюджетна образователна институция за висше образование "Ростовски държавен медицински университет" на Министерството на здравеопазването на Русия (списание "Неврология, невропсихиатрия, психосоматика" " № 9(4), 2017) [прочети];
статия „Митохондриални цитопатии: синдроми MELAS и MIDD. Един генетичен дефект - различни клинични фенотипове” Муранова А.В., Строков И.А.; Федерална държавна бюджетна образователна институция за висше образование „Първи Московски държавен медицински университет на името на. ТЯХ. Сеченов“ Министерство на здравеопазването на Руската федерация, Москва (Неврологичен журнал, № 1, 2017 г.) [прочетете];
статия „Инсулт-подобни епизоди при митохондриална енцефаломиопатия с лактатна ацидоза“ L.A. Калашникова, Л.А. Добринина, А.В. Сахарова, Р.П. Чайковская, М.Ф. Мир-Касимов, Р.Н. Коновалов, А.А. Шабалина, М.В. Костирева, В.В. Гнездицки, С.В. Процки; Научен център по неврология на Руската академия на медицинските науки, Москва (списание „Анали по клинична и експериментална неврология“ № 3, 2010) [прочетете];
статия „Неврологични нарушения при митохондриална енцефаломиопатия - лактатна ацидоза с инсулт-подобни епизоди (MELAS синдром)” от D.A. Харламов, А.И. Крапивкин, В.С. Сухоруков, Л.А. Куфтина, О.С. Грознова; Московски научноизследователски институт по педиатрия и детска хирургия (списание „Руски бюлетин по перинатология и педиатрия“ № 4 (2), 2012 г.) [прочетете];
статия „Инсултоподобен курс на митохондриална енцефаломиопатия (MELAS синдром)“ от I.N. Смирнова, Б.А. Кистенев, М.В. Кротенкова, З.А. Суслина; Научен център по неврология на Руската академия на медицинските науки, Москва (списание "Нервни болести" № 1, 2006 г.) [прочетете];
статия „Инсулти при митохондриални заболявания” N.V. Пизова, Катедра по нервни болести с курсове по неврохирургия и медицинска генетика, Ярославска държавна медицинска академия (списание „Неврология, невропсихиатрия, психосоматика” № 9(4), 2017) [прочетете];
статия „Исхемичен инсулт като проява на митохондриална енцефалопатия при млад пациент“ Murzaliev A.M., Lutsenko I.L., Musabekova T.O., Akbalaeva B.A. (списание „Наука и нови технологии” № 6, 2011) [прочетете];
статия „Епилепсия при синдром на MELAS“ Мухин К.Ю., Миронов М.Б., Никифорова Н.В., Михайлова С.В., Чадаев В.А., Алиханов А.А., Рижков Б.Н., Петрухин А.С.; Държавна образователна институция за висше професионално образование RSMU Roszdrav; Руска детска клинична болница (Руски журнал по детска неврология" № 3, 2009 г.) [прочетете];
статия „Алгоритъм за диагностициране на митохондриални енцефаломиопатии“ от S.N. Илариошкин, Изследователски институт по неврология на Руската академия на медицинските науки (списание "Нервни болести" № 3, 2007 г.) [прочетете]
© Laesus De Liro
Синдромът на MELAS е митохондриално заболяване, характеризиращо се с увреждане на мускулите и централната нервна система.
MELAS (англ. Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes - “митохондриална енцефаломиопатия, лактатна ацидоза, stroke-like episodes”) е прогресивно невродегенеративно заболяване, характеризиращо се с проявите, изброени в името, и е придружено от полиморфни симптоми - инсулт , диабет, гърчове, намален слух, сърдечни заболявания, нисък ръст, ендокринопатии, непоносимост към упражнения и невропсихиатрични разстройства.
История.
Синдромът MELAS е описан за първи път през 1984 г. от Pavlakis и колеги; десет години по-късно Павлакис и Мицио Хирано публикуват преглед на 110 случая.
Тип наследяване:
майчина
Епидемиология:
Точната честота на заболяването не е известна. В литературата има ограничени данни за честотата на заболяването. В Северна Финландия честотата на мутацията A3243G е 16,3:100 000.
Патогенеза:
Мутациите на митохондриалната ДНК, които контролират дихателната верига на митохондриите, са придружени от нарушаване на процесите на окислително фосфорилиране, най-важният източник на енергия за метаболитните процеси в клетката.
Клинични проявления
Под 40-годишна възраст се приемат пациенти с MELAS с преходна исхемична атака, както и с епилепсия, многократно повръщане, главоболие и мускулна слабост. Тези пациенти често са клинично диагностицирани с деменция.
Младата възраст и липсата на рискови фактори, характерни за инсулт, помага да се мисли за MELAS.
Лабораторни данни
Лактатната ацидоза е повишаване на нивата на лактат и пируват.
Данни за визуализация
Промените в мозъка са подобни на тези, причинени от инсулт.
Разлики от инсулт
1) засегнатите области не съвпадат с границите на артериалните съдови територии.
2) при повтарящи се атаки лезиите се визуализират на различно място.
+ клинични данни (млада възраст, липса на рискови фактори за инсулт).
CT
Множество хиподенсни области, които не съответстват на съдовата територия.
Калцификация на базалните ганглии (най-често при по-възрастни пациенти).
Атрофията възниква на фона на регресия и клинично подобрение.
ЯМР
Остър миокарден инфаркт
За разграничаване от инсулт се използват ADC и DWI (при инсулти дифузията е ограничена (цитотоксичен оток), а при MELAS дифузията е ограничена леко или без промени (вазогенен оток).
Включване в патологичния процес на субкортикалното бяло вещество на мозъка.
Влошаване на визуализацията на яснотата на контурите на гирусите и увеличаване на сигнала от тях на Т2-претеглени изображения.
Хроничен инфаркт
Промените могат да бъдат симетрични или асиметрични.
Фокалната атрофия възниква на фона на регресия и клинично подобрение.
Най-често се засягат париеталните, тилните и темпоралните дялове на мозъка.
MR спектроскопия
Повишени нива на лактат.

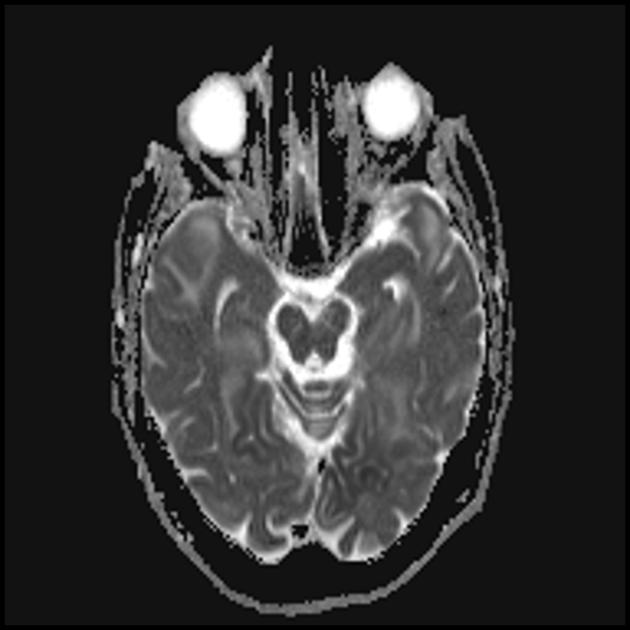

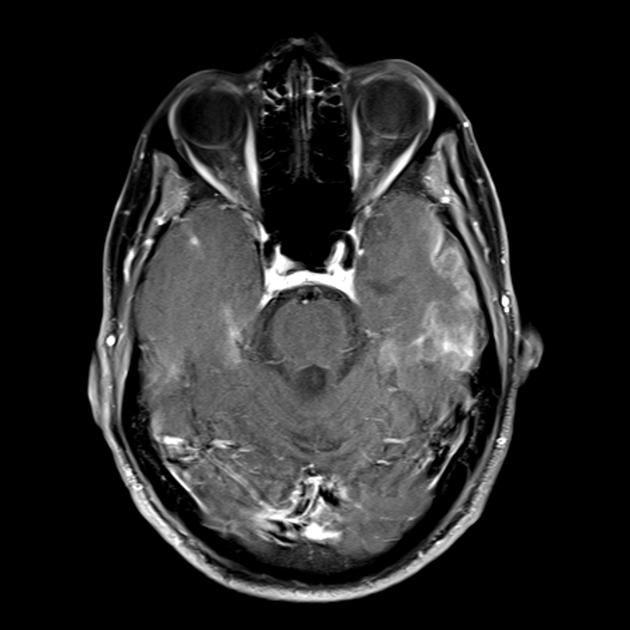
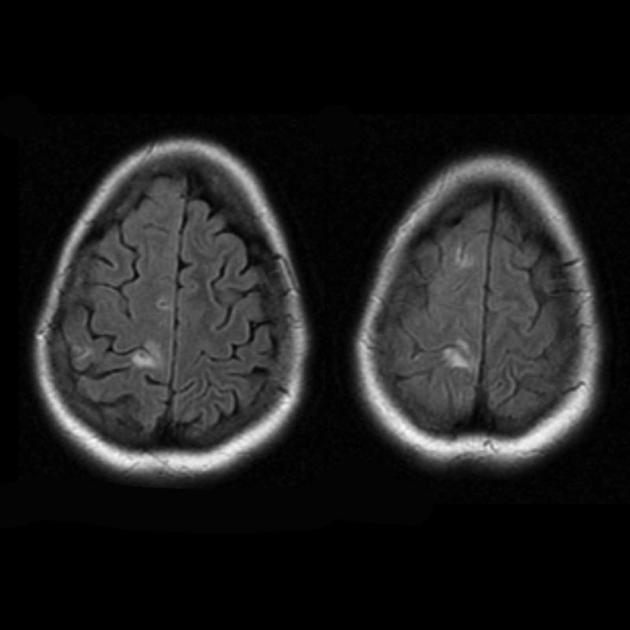

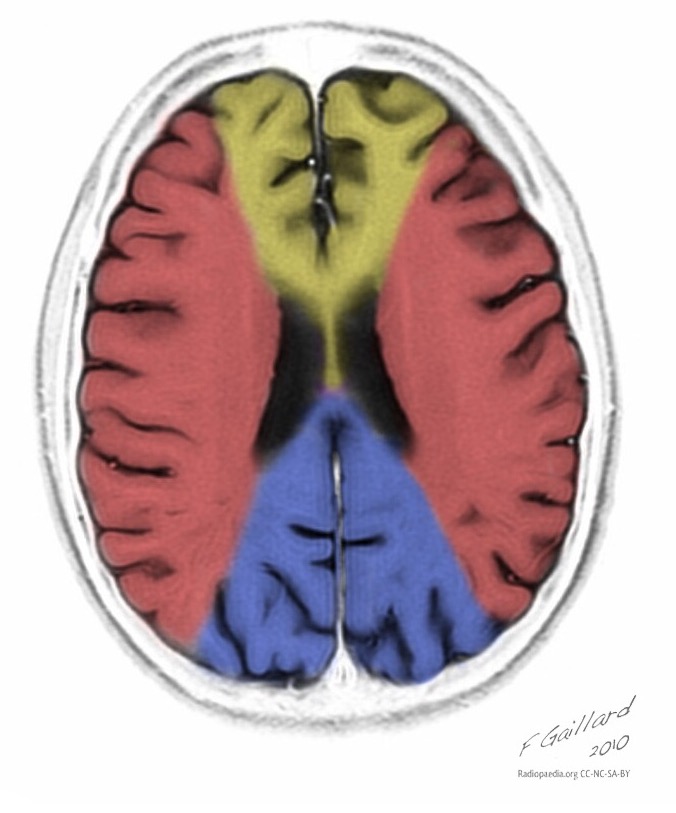
Синдромът на MELAS (митохондриална енцефаломиопатия, лактатна ацидоза и инсулт) - митохондриална енцефаломиопатия, лактатна ацидоза и инсулт - е изолиран сравнително наскоро (през 1984 г.). Заболяването е свързано с точкова мутация в митохондриалната ДНК, която е 90% локализирана в гена, кодиращ синтеза на левцинова трансферна РНК, което предотвратява включването му в протеините на дихателната верига. Както при всички митохондриални заболявания, диагнозата на синдрома на MELAS е трудна поради значителната вариабилност в клиничната изява.
Основните клинични прояви на синдрома MELAS са: непоносимост към физическо натоварване; повтарящи се състояния, подобни на инсулт. Терминът „инсултоподобен” вероятно се дължи на факта, че в повечето от описаните случаи водеща клинична изява е главоболие с повръщане, конвулсии, често с нарушено съзнание, с продължителност от няколко часа до няколко дни.
По време на тези атаки редица пациенти често развиват неврологични разстройства под формата на хемианопсия, хемипареза и рядко под формата на афазия. При извършване на компютърна томография 60% от тези пациенти разкриват огнища на ниска плътност и полиморфни гърчове; биохимично изследване разкрива лактатна ацидоза; начало на заболяването - на 5-6 годишна възраст; ходът на заболяването е прогресивен.
Представяме собствено наблюдение на синдрома MELAS.
Пациент А., на 6 години, е приет в клиниката с оплаквания от остро развита слабост в десните крайници, продължила няколко часа, и главоболие. Роден от първа бременност. Бременността и раждането са протекли без усложнения. Ранното психомоторно развитие на детето е съобразено с възрастта. От 1 година до 2 години са отбелязани афективно-респираторни пароксизми. От 3-годишна възраст се развиват чести ацетонемични състояния на върха на главоболието.
По време на прегледа в клиниката: двигателно разстройство, безпокойство, умора при стрес (психически и физически). Неврологичен статус: черепна инервация без характеристики, мускулна хипотония, асиметрия на сухожилните рефлекси (по-ярки вдясно). Няма парези. Няма атаксия.
По време на първоначалния ЯМР на мозъка в левия тилен дял се открива голяма зона на хиперинтензивен сигнал в режим Т2 и хипоинтензивен в режим Т1 с ясни контури, средните структури не са изместени. Тази радиологична картина се разглежда като исхемичен инсулт.
През следващата година детето получава три пъти парциални епилептични припадъци и повтарящи се епизоди на пароксизмално главоболие с повръщане. Получава антиконвулсивна терапия. Година след първото лечение в клиниката, момчето е прието отново поради пристъп на генерализирани тонично-клонични гърчове, появили се два пъти в рамките на един час.
Летаргията и слабостта се увеличават, появява се пароксизмално главоболие с еднократно повръщане. ЯМР на мозъка се повтаря - в режимите Т1 и Т2 се визуализират зони с променен сигнал в теменно-тилната област от двете страни (а лезията вляво е с по-малък размер в сравнение с тази при предишното изследване с ЯМР). Така за няколко месеца след първия инсулт момчето претърпя поне още три остри мозъчно-съдови инцидента.
Метаболитните нарушения се изразяват в значително повишаване на нивото на лактат в кръвта до 4,2 mmol/l (норма до 1,7 mmol/l) и пируват.
Цялостният анализ на прегледите и клиничните резултати позволи да се установи специфична нозологична форма на митохондриална енцефаломиопатия - синдром на MELAS, който беше установен в клиниката за първи път.
Впоследствие детето получава антиконвулсивна терапия (Топамакс 5 mg/kg/ден), лечение, насочено към стимулиране на тъканното дишане с коензим Q-10, янтарна киселина, Cytomac, както и Actovegin и Contrical.
Даденият случай на рецидивиращи метаболитни мозъчни инфаркти при дете, причинени от MELAS синдром, не е единственият случай в клиниката и се натрупва база данни от подобни пациенти.
Материалите са предназначени за невролози, терапевти и общопрактикуващи лекари.
Сергей Лихачов, ръководител, доктор по медицина. науки, професор;
Инеса Плешко, водещ изследовател, кандидат на медицинските науки. Науки, неврологично отделение на Републиканския научно-практически център по неврология и неврохирургия.
Церебралната автозомно доминантна артериопатия с подкорови инфаркти и левкоенцефалопатия (CADASIL) е прогресивно автозомно доминантно заболяване, чиито клинични прояви включват повтарящи се субкортикални исхемични инсулти, мигрена, субкортикална деменция и афективни разстройства. Текуща заболеваемост - 1 случай
на 100 000 души население.
В Републиканския научно-практически център по неврология и неврохирургия се наблюдават 7 пациенти (от тях 4 жени) с CADASIL; възраст - от 32 до 68 години. Изследвани са с неврологични и молекулярно-генетични методи. Имаше характерни симптоми; анамнеза за мигрена, повтарящи се лакунарни инсулти и афективни разстройства. ЯМР на мозъка разкрива подкорови инфаркти и левкоенцефалопатия, характерни за CADASIL.
При 2 души молекулярно-генетичната диагностика разкри хетерозиготна мутация в гена Notch3 на хромозома 19, която причинява CADASIL. Notch гените кодират трансмембранни рецептори, участващи в клетъчната онтогенеза. С CADASIL в повечето случаи се определят миссенс мутации, поради което се променя структурата на трансмембранния протеин и се нарушават неговите функции.
Патогенезата на CADASIL не е напълно ясна. Смята се, че основният фактор е артериопатията с прогресираща оклузия на малки перфориращи съдове на бялото вещество на мозъка (водещи до хронична хипоперфузия). В този случай се откриват характерни гранулирани осмиофилни включвания, причиняващи пролиферация на компонентите на базалната мембрана, удебеляване на tunica media и механична компресия на малките артерии. В резултат на това кръвно-мозъчната бариера се уврежда и се развива оток.
Допълнителен патологичен фактор е активирането на астроцити в близост до съдовата стена. Те освобождават ендотел-1, причинявайки вазоконстрикция и нарушен кръвен поток.
Съставът на гранулираните осмиофилни включвания е неизвестен. Предполага се, че протеинът Notch3 е един от техните компоненти. В кожни биопсии на пациенти с Notch3 мутация, осмиофилни гранули и дегенерация на гладкомускулни клетки могат да бъдат открити преди 20-годишна възраст.
Клинична диагноза на CADASIL:
- семейна история;
- развитие на първите симптоми на заболяването преди 50-годишна възраст;
- наличието на два от следните симптоми - мигрена, повтарящи се инсулти, разстройства на настроението, субкортикална деменция.
Трябва да се изключат съдови рискови фактори, етиологично свързани с неврологични симптоми. ЯМР показва лезии на бялото вещество на мозъчните хемисфери и липса на кортикални инфаркти.
Надеждната диагноза на CADASIL се потвърждава от положителен резултат от молекулярно-генетична диагностика или откриване на артериопатия с характерни гранулирани осмиофилни включвания по време на кожна или мускулна биопсия.
Най-честите симптоми на CADASIL са преходни исхемични атаки и исхемични инсулти, наблюдавани при почти 85% от пациентите.
Те се характеризират с рецидивиращ курс, проявяващ се като синдром на класически лакунарен инсулт и пълна клинична ремисия след няколко дни или седмици.
На второ място по честота са когнитивните увреждания (отбелязани при 60% от пациентите). Те могат да започнат на 35-годишна възраст, понякога дори преди исхемични епизоди. Приблизително 75% от случаите с CADASIL развиват деменция. Първият симптом обикновено е мигрена; често се появява преди 20-годишна възраст и обикновено предшества инсулти.
Данните за участието на сърцето в патологичния процес при CADASIL са противоречиви. L. Oberstein и др. (2003) установяват, че 25% от пациентите, диагностицирани с CADASIL, са имали анамнеза за остър миокарден инфаркт или аномалия на Q-зъбеца на електрокардиограмата. В друго проучване Cumurciuc et al. (2006) не откриха положителна сърдечна история при 23 души с мутация в гена Notch3.
Клиничните прояви на CADASIL и церебралната микроангиопатия с друга етиология са сходни - необходима е диференциална диагноза.
За своевременно идентифициране на CADASIL при пациенти и членове на техните семейства е необходимо да се прибегне до молекулярно-генетични методи и/или хистологични изследвания.
MELAS синдром
Митохондриалната енцефаломиопатия с лактатна ацидоза и удароподобни епизоди (MELAS) е рядко наследствено заболяване, причинено от патология на митохондриалния геном, нарушаване на енергийния метаболизъм и функционирането на най-енергийно зависимите органи и тъкани (ЦНС, сърдечни и скелетни мускули, очи, бъбреци, черен дроб, костен мозък, ендокринна система). Широката вариабилност на клиничните прояви на синдрома MELAS и неговата рядка поява предопределят трудности при диагностицирането на практикуващия лекар.
В Републиканския научно-практически център по неврология и неврохирургия се наблюдават 3 пациенти (жена на 46 години и нейните синове на 24 и 23 години) с диагноза MELAS синдром. Извършени са им клиничен неврологичен преглед, молекулярно-генетична диагностика и ЯМР на мозъка.
Всички са ниски; анамнеза за симптоми на митохондриална патология: сензоневрална загуба на слуха, мигреноподобни главоболия, лоша толерантност към упражнения. Началото на заболяването е генерализирани конвулсивни припадъци. При 2 пациенти първите симптоми са се появили преди 20-годишна възраст; имаше епилептични припадъци, следващи един след друг, епизоди на зрително увреждане с наличие на огнища на невроизобразяване в тилната и темпоралната област, повишени нива на лактат в кръвта и цереброспиналната течност. Един човек показа умерен спад в когнитивната функция; според ултразвук на сърцето - хипертрофична кардиомиопатия; диабет.
Молекулярно-генетично изследване разкрива мултисистемни лезии, типични за MELAS, широка вариабилност и различни степени на тежест на клиничните прояви, съответстващи на броя на мутантните копия на A3243G в гена tRNALeu(UUR).
MELAS се характеризира с майчин тип наследство, наличието на спорадични случаи, когато възниква мутация de novo; натрупване в клетки - нормални и мутантни типове - на митохондриална ДНК (хетероплазмия) и произволно разпределение по време на деленето между дъщерните клетки (митотична сегрегация). На генетично ниво причината за синдрома на MELAS е хетероплазменото пренареждане 3243A>G в гена tRNALeu(UUR) (открива се в 80% от случаите).
Патогенезата на заболяването все още не е проучена. Има 2 основни теории - „митохондриална ангиопатия” и „митохондриална цитопатия”. Известно е, че удароподобните лезии не съответстват на съдовите зони и се разпространяват в околните области поради съпътстващ вазогенен оток, причинен от продължителна епилептична активност. Смята се, че епизодите, подобни на инсулт, са причинени от неврална свръхвъзбудимост в локализирана област на мозъка. Възниква от митохондриална дисфункция в капилярни ендотелни клетки, неврони или астроцити; деполяризира съседните неврони, което води до разпространение на епилептична активност.
В допълнение, в интервалите между епизодите, подобни на инсулт, еднофотонна емисионна компютърна томография (SPECT) показва, че пациентите с MELAS имат хипоперфузия на задния цингулатен кортекс, което показва нарушение на церебралната хемодинамика.
Нарушеното окислително фосфорилиране и разрушаването на митохондриалната респираторна верига допринасят за преобладаването на катаболитния метаболизъм и промените от цикъла на Кребс към анаеробна гликоза с натрупване на лактат. Високите нива на последните в централната нервна система обикновено корелират с периоди на неврологични симптоми.
Основните клинични признаци на MELAS са епизоди, подобни на инсулт, лактатна ацидоза и наличието на „накъсани червени влакна“ в мускулни биопсии. Допълнителни прояви могат да включват деменция, психоза, епилептични пароксизми, мигреноподобни главоболия, атаксия, миопатия, калцификация на базалните ганглии според невроизобразяване, оптична атрофия, ретинопатия, глухота, диабет, чревна псевдообструкция, кардиомиопатия.
Ранната възраст на поява на MELAS е от 5 до 20 години, но има наблюдения за късно начало - през 5-6-то десетилетие от живота. Има случаи, когато синдромът започва след сърдечни заболявания.
Мултисистемният характер на лезията при MELAS усложнява клиничната диагноза.
Наследственият характер на заболяването налага извършването на молекулярно-генетични изследвания за поставяне на точна диагноза.
и идентифицира други пациенти сред роднините на пациента.
Материалите са предназначени за невролози, терапевти и общопрактикуващи лекари.
 (1оценки, средно: 5,00от 5)
(1оценки, средно: 5,00от 5)