Melasse-Syndrom. Seltene Krankheiten. Text der wissenschaftlichen Arbeit zum Thema „Epilepsie beim Melas-Syndrom“
Unter dem MELAS-Syndrom versteht man mitochondriale Erkrankungen (MD), die durch genetische und strukturell-biochemische Defekte der Mitochondrien verursacht werden und mit einer gestörten Gewebeatmung und infolgedessen einem systemischen Defekt des Energiestoffwechsels einhergehen, wodurch die meiste Energie verbraucht wird -abhängige Gewebe und Zielorgane sind in verschiedenen Kombinationen betroffen: Gehirn, Skelettmuskulatur und Myokard, Bauchspeicheldrüse, Sehorgan, Nieren, Leber. Klinische Störungen dieser Organe können in jedem Alter auftreten. Gleichzeitig erschwert die Heterogenität der Symptome die klinische Diagnose dieser Erkrankungen. Die Notwendigkeit, MB auszuschließen, ergibt sich bei Vorliegen multisystemischer Manifestationen, die nicht in den üblichen pathologischen Prozess passen. Die Häufigkeit einer Funktionsstörung der Atmungskette wird auf 1 von 5 bis 10.000 bis 4 bis 5 pro 100.000 Neugeborene geschätzt.
lesen Sie auch den Beitrag: Mitochondriale Erkrankungen(zur Website)
Das MELAS-Syndrom (Mitochondriale Enzephalomyopathie, Laktatazidose und schlaganfallartige Episoden) ist eine Multisystemerkrankung, die durch schlaganfallartige Episoden gekennzeichnet ist, die in jungen Jahren (vor dem 40. Lebensjahr) auftreten, Enzephalopathie mit Anfällen und Demenz sowie mitochondriale Myopathie mit dem Phänomen „ „zackige“ rote Fasern und Laktatazidose (ein Anstieg des Milchsäurespiegels im Blut ohne Azidose ist möglich).
Das MELAS-Syndrom wird durch Punktmutationen in der mitochondrialen DNA (mtDNA) verursacht. Die Krankheit wird mütterlicherseits vererbt (daher sind Verwandte mütterlicherseits wahrscheinlich Träger solcher Mutationen; häufiger beschreiben Verwandte mütterlicherseits ein oligosymptomatisches Krankheitsbild mit einzelnen Symptomen des MELAS-Syndroms; bei asymptomatischen Verwandten ist es das MELAS-Syndrom nur durch die Ergebnisse einer Muskelbiopsie oder molekularer Forschung identifiziert). Derzeit sind mehr als zehn Gene bekannt, deren Mutationen zur Entstehung des Krankheitsbildes MELAS-Syndrom führen. In den meisten Fällen wird die Entwicklung des MELAS-Syndroms durch Mutationen in Genen verursacht, die für die Funktionen der Transfer-RNA kodieren.
Typischerweise beginnt die Krankheit im Alter zwischen 6 und 10 Jahren (das Erkrankungsalter liegt zwischen 3 und 40 Jahren; ein früher Ausbruch der Krankheit ist typisch und tritt bei 90 % der Patienten auf). Die Patienten zeichnen sich durch Kleinwuchs (und Belastungsintoleranz) aus. Seitens der inneren Organe können Kardiomyopathie, Reizleitungsstörungen des Herzens, Diabetes mellitus, Nephropathie und Motilitätsstörungen des Magen-Darm-Traktes beobachtet werden.
Erinnern! Die wichtigsten klinischen Kriterien für die Diagnose von MELAS sind: [ 1 ] mütterliche Art der Vererbung; [ 2 ] Beginn vor dem 40. Lebensjahr; [ 3 ] normale psychomotorische Entwicklung vor der Krankheit; [ 4 ] Belastungsunverträglichkeit; [ 5 ] migräneähnlicher Kopfschmerz mit Übelkeit und Erbrechen; [ 6 ] Schlaganfall-ähnliche Episoden; [ 7 ] Enzephalopathie mit epileptischen Anfällen und/oder Demenz (myoklonische Anfälle werden am häufigsten registriert, aber auch fokale sensorische, motorische und sekundär generalisierte tonisch-klonische Anfälle werden beobachtet); [ 8 ] Laktatazidose; [ 9 ] ausgefranste rote Fasern in Skelettmuskelbiopsien; [ 10 ] progressiver Kurs.
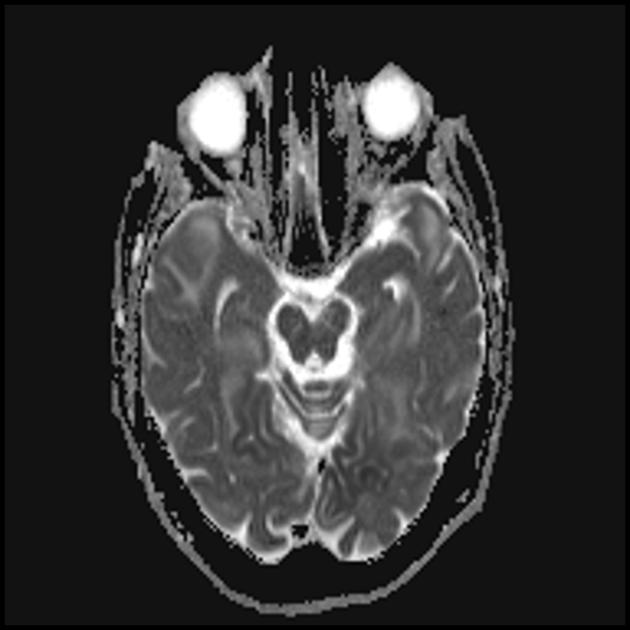
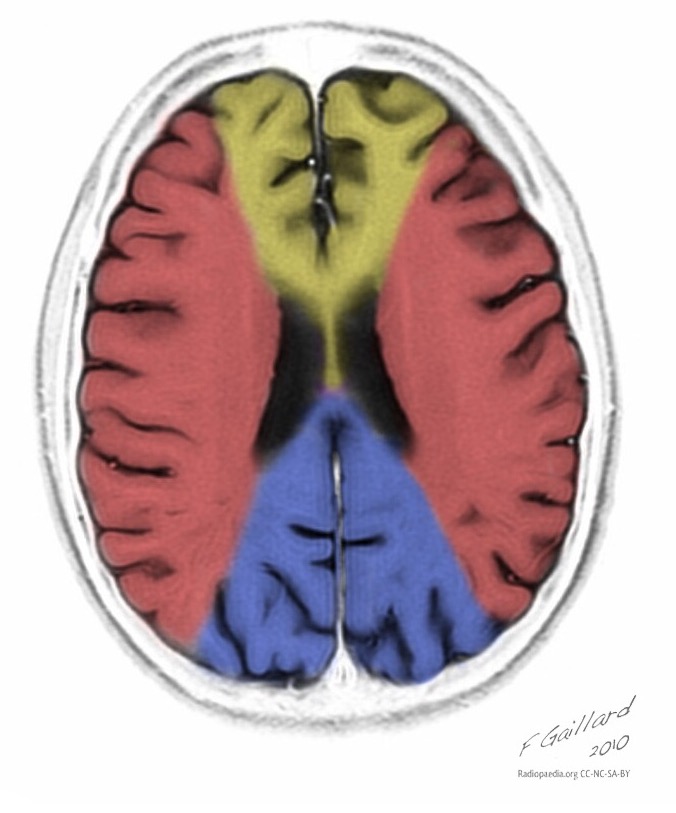
Das charakteristische klinische Merkmal des MELAS-Syndroms sind schlaganfallähnliche Episoden (IPE), die die plötzliche Entwicklung fokaler neurologischer Störungen verursachen. Ein charakteristisches Merkmal der IPE ist die „hintere“ Lokalisierung von Läsionen im Gehirn. Am häufigsten befinden sich Läsionen im Okzipital-, Parietal- und Temporallappen, seltener im Frontallappen, im Kleinhirn oder in den Basalganglien; oft sind es mehrere. Die Selektivität der Lokalisierung fokaler Veränderungen bestimmt die Merkmale fokaler neurologischer Symptome: Hemianopsie, sensorische Aphasie, Akalkulie, Agraphie, optisch-räumliche Störungen, Ataxie, Bewusstseinsveränderungen ([ !!! ] Am häufigsten sind die Läsionen in der Kortikalis der Hinterhauptslappen der Großhirnhemisphären lokalisiert, was zu Hemianopsie oder kortikaler Blindheit führt. Schlaganfälle können in Form von klinischen und/oder radiologischen Veränderungen (abhängig von der Schwere der durch Energiemangel der Neuronen verursachten Stoffwechselstörungen) verschwinden oder langfristig festgestellt werden. In symmetrischen Bereichen kommt es häufig im Abstand von 1 bis 3 Monaten zu wiederholten „Hirninfarkten“. Diese Läsionen können klein oder groß, einzeln oder mehrfach sein, sie sind meist asymmetrisch und ihre Lokalisation entspricht nicht dem Bereich der Blutversorgung. Darüber hinaus können bei Patienten mit MELAS-Syndrom Verkalkungen in den Basalganglien auftreten (in diesen Fällen kann eine CT-Untersuchung des Gehirns hilfreich sein). Im neurologischen Status äußern sich diese morphologischen Veränderungen durch Myoklonien, Ataxie, Episoden akuter Psychosen oder Bewusstseinsstörungen (Defizite) bis hin zum Koma ([ !!! ] Merkmal dieser akuten Episoden, inkl. Schlaganfälle, einerseits eine schnelle [von mehreren Stunden bis zu mehreren Wochen] Rückbildung der Symptome, andererseits eine Rückfallneigung); von den Sinnesorganen werden Atrophie der Sehnerven, Pigmentretinopathie und Hörverlust festgestellt.
Es wird angenommen, dass die folgenden Mechanismen bei der Entstehung von IPE wichtig sind: [ 1 ] Stoffwechselstörungen im Gehirn mit der Entwicklung einer Laktatazidose aufgrund eines mitochondrialen Energiemangels; [ 2 ] zerebrale Ischämie, verursacht durch mitochondriale Angiopathie auf der Ebene kleiner Arterien; [ 3 ] Lokale Erhöhung der neuronalen Erregbarkeit aufgrund einer mitochondrialen Dysfunktion in Neuronen, Astrozyten oder Kapillarendothel, die sich allmählich in der gesamten Großhirnrinde ausbreitet, geht mit der Entwicklung von Ödemen einher und kann zu laminarer Nekrose in der Großhirnrinde führen.
Auf den ersten Blick ähnelt ein Schlaganfall mit MELAS-Syndrom einem normalen Schlaganfall aufgrund einer Thrombose oder Embolie. Tatsächlich sind schlaganfallartige Episoden beim MELAS-Syndrom untypisch: Sie treten bei jungen Menschen auf, werden häufig durch Infektionskrankheiten hervorgerufen und können in Form von migräneähnlichen Kopfschmerzen oder Krampfanfällen auftreten. Die MRT-Untersuchung einer akuten IPE beim MELAS-Syndrom zeigt Anomalien wie ein erhöhtes Signal auf T2-gewichteten oder FLAIR-Bildern (Water-Attenuated Inversion Recovery). Die Läsionen fallen nicht mit den Territorien der großen Hirnarterien zusammen, sondern betreffen größtenteils den Kortex und die darunter liegende weiße Substanz, mit mäßiger Beteiligung der tiefen weißen Substanz. Akute Hirnläsionen im MRT beim MELAS-Syndrom können sich verändern, wandern oder sogar verschwinden ([ !!! ] gekennzeichnet durch Fluktuation der Herde, bestimmt durch MRT). Die Angiographie zeigt das Fehlen einer signifikanten Gefäßpathologie: Zusätzlich zu den normalen Ergebnissen kann eine Vergrößerung des Arterien- und Venenkalibers oder eine Kapillarhyperämie festgestellt werden.
Neuromorphologische Untersuchungen des Gehirns beim MELAS-Syndrom zeigen das Vorhandensein einer multifokalen Nekrose, die hauptsächlich in der Großhirnrinde und der subkortikalen weißen Substanz sowie im Kleinhirn, Thalamus und Basalganglien lokalisiert ist. Die Läsionen ähneln Infarktbereichen, decken sich aber, wie oben erwähnt, nicht mit den Becken großer Hirngefäße. Außerdem kommt es zu einer spongiformen Degeneration der Großhirnrinde, einer Vermehrung der Kapillaren und einer Erschöpfung der Neuronen.
Erinnern! Schlaganfallähnliche Episoden bei MELAS weisen folgende Merkmale auf: [ 1 ] junges Alter (normalerweise bis zu 40 Jahre); [ 2 ] häufiges Vorliegen eines auslösenden Faktors (tritt nach fieberhaftem Fieber, epileptischem Anfall, migräneähnlichen Kopfschmerzen auf); [ 3 ] Lieblingslokalisation – Hinterhauptsregion; [ 4 ] Läsionen befinden sich in der Regel außerhalb des Bereichs großer Hirnarterien, am häufigsten in der Kortikalis oder in tiefen Strukturen der weißen Substanz des Gehirns.
Bei der Differentialdiagnose von IPE und Hirninfarkt werden folgende Symptome berücksichtigt:
■ allmähliche, über mehrere Tage verlaufende Zunahme fokaler neurologischer Symptome (die pathophysiologische Grundlage für dieses Entwicklungstempo ist die allmähliche Zunahme des Energiemangels des Gehirns aufgrund einer gestörten oxidativen Phosphorylierung in Mitochondrien);
■ Eine allmähliche Abnahme des Wachzustands, die im Widerspruch zu einem relativ leichten fokalen neurologischen Defizit steht, geht nicht mit einem sekundären Hirnstammsyndrom einher und kann daher nicht durch eine Zunahme von Hirninfarkten und Ödemen erklärt werden (diese Symptome sind ebenfalls vorhanden). basierend auf einer Stoffwechselstörung, die durch eine Verletzung der Energieversorgung des Gehirns verursacht wird);
■ Entwicklung in der akuten Phase wiederholter lokaler und generalisierter epileptischer Anfälle, die laut Literatur bei 2/3 der Patienten mit IPE auftreten (Anfälle sind nicht mit einem zerebrovaskulären Unfall verbunden, da die Quelle ihrer Entstehung in beiden Fällen die Entladungsaktivität ist). Hemisphären des Gehirns und nicht in Strukturen, die auf ein bestimmtes Becken von Hirnarterien beschränkt sind; die wiederkehrende Natur von Anfällen und das Fehlen ausgeprägter anhaltender fokaler neurologischer Symptome sind ebenfalls nicht charakteristisch für einen akuten Hirninfarkt);
■ vollständige Durchgängigkeit der Hirnarterien gemäß Duplex-Scan der brachiozephalen Arterien und zerebraler Angiographie, was für einen ischämischen Schlaganfall nicht typisch ist;
■ Merkmale des Neuroimaging-Bildes: vorwiegend kortikale Lokalisierung von Herden und ihre „hintere Lage“, was charakteristisch für MELAS ist und durch die größere Anfälligkeit von Neuronen in diesen Bereichen aufgrund ihres größeren Energiebedarfs erklärt wird; Ein weiteres Merkmal der Neurobildgebung ist das Verschwinden einiger Herde, das offenbar auf einem Ödem und nicht auf einer Nekrose der Gehirnsubstanz aufgrund von Stoffwechselstörungen beruht.
![]()
MELAS-Syndrom An RADIOPÄDIE.org
Eine der Haupterscheinungen des MELAS-Syndroms ist auch Muskelschwäche (myopathisches Syndrom). Die Unspezifität dieses Symptoms lässt jedoch keine Diagnose zu. Erst wenn Migräne, Krampfanfälle und/oder Schlaganfall-ähnliche Ereignisse auftreten, kann der Ausbruch eines MELAS-Syndroms diagnostiziert werden.
Screening-Tests für das MELAS-Syndrom sind Neuroimaging und eine Untersuchung des Laktatspiegels [Anstieg] im Blut (teilweise in der Gehirn-Rückenmarks-Flüssigkeit) – ein Bluttest auf Milchsäure (Laktat) und Brenztraubensäure (Blutlaktatspiegel [normal] – venöses Blut). - 0,5 - 2,2 mmol/l, arterielles Blut - 0,5 - 1,6 mmol/l; Laktat/Pyruvat-Verhältnis - 10/1). Die Diagnose kann durch DNA-Tests bestätigt werden, um die häufigsten Mutationen zu bestimmen. Liegen beim MELAS-Syndrom keine häufigen Punktmutationen vor, kann eine Muskelbiopsie (unter Verwendung der Dreifarbenmethode von Gomori zum Nachweis von Ragged Red Fibers [RRFs] – Myofibrillen mit einem hohen Anteil des mutierten Genoms und einer großen Anzahl proliferierender veränderter Mitochondrien) hilfreich sein bei der Diagnose. Mithilfe der Biopsie lässt sich auch das Vorhandensein biochemischer Defekte in der Atmungskette feststellen, die hauptsächlich mit den Enzymen Succinatdehydrogenase und Cytochromoxidase zusammenhängen.

Die Behandlung des MELAS-Syndroms umfasst zwei Hauptbereiche. Die erste ist die Syndromtherapie (der Schwerpunkt liegt auf Epilepsie, Diabetes usw.). Es unterscheidet sich nicht von allgemein anerkannten Ansätzen zur Behandlung von Syndromen. Das Stoppen epileptischer Anfälle ist notwendig, da der Stoffwechselstress, der während der Anfälle auftritt, die Entwicklung schlaganfallähnlicher Episoden auslösen kann. Valproinsäure-Derivate, die in der Epileptologie weit verbreitet sind, hemmen die Mitochondrienfunktionen und ihre Verwendung ist unerwünscht. Wenn ein Absetzen des Arzneimittels nicht möglich ist, sollten Sie gleichzeitig Levocarnitin in einer Dosis von bis zu 100 mg/kg pro Tag einnehmen. Auch Phenytoin und Barbiturate sollten gemieden werden. Die zweite Behandlungsrichtung ist pathogenetisch, derzeit gibt es jedoch keine wirksame pathogenetische Therapie. Die Behandlungsstrategie zielt auf die Verbesserung des Energiestoffwechsels der Zelle ab und umfasst die Gabe von Coenzym Q oder Idebinon (Noben), Bernsteinsäurepräparaten, Vitamin K1 und K3, Nicotinamid, Riboflavin, L-Carnitin, Antioxidantien (Mexidol, Mildronat, Vitamine). E und C), Laktatkorrektoren – Azidose (Dimephosphon). [ !!!
] Der Einsatz von Medikamenten, die die Mitochondrienfunktion hemmen (Barbiturate, Valproate, Statine, Glukokortikoide), ist zu vermeiden.
Lesen Sie mehr über das MELAS-Syndrom in den folgenden Quellen:
Präsentation „MELAS-Syndrom“ Kuzenkova L.M., Globa O.V.; Abteilung für Psychoneurologie, Forschungsinstitut für Pädiatrie, Wissenschaftliches Zentrum für Kindergesundheit, Russische Akademie der Medizinischen Wissenschaften, Moskau [lesen];
Artikel „Mitochondriale Enzephalopathie mit Schlaganfall-ähnlichen Episoden und Laktatazidose (MELAS-Syndrom): Diagnosekriterien, Merkmale epileptischer Anfälle und Behandlungsansätze am Beispiel eines klinischen Falles“ Yamin M.A., Chernikova I.V., Araslanova L.V., Shevkun P.A.; Staatliche autonome Einrichtung der Region Rostow „Regionales Beratungs- und Diagnosezentrum“; Abteilung für Neurologie und Neurochirurgie mit Kursen für manuelle Therapie und Reflexzonenmassage der Fakultät für Bildung und Ausbildung der föderalen staatlichen Bildungseinrichtung „Staatliche Medizinische Universität Rostow“ des Gesundheitsministeriums Russlands (Zeitschrift „Neurologie, Neuropsychiatrie, Psychosomatik“) " Nr. 9(4), 2017) [lesen];
Artikel „Mitochondriale Zytopathien: MELAS- und MIDD-Syndrome. Ein genetischer Defekt – verschiedene klinische Phänotypen“ Muranova A.V., Strokov I.A.; Staatliche Haushaltsbildungseinrichtung für höhere Bildung „Erste Moskauer Staatliche Medizinische Universität, benannt nach. IHNEN. Sechenov“ Gesundheitsministerium der Russischen Föderation, Moskau (Neurologische Zeitschrift, Nr. 1, 2017) [lesen];
Artikel „Schlaganfallartige Episoden bei mitochondrialer Enzephalomyopathie mit Laktatazidose“ L.A. Kalashnikova, L.A. Dobrynina, A.V. Sacharowa, R.P. Tschaikowskaja, M.F. Mir-Kasimov, R.N. Konovalov, A.A. Shabalina, M.V. Kostyreva, V.V. Gnezditsky, S.V. Protsky; Wissenschaftliches Zentrum für Neurologie der Russischen Akademie der Medizinischen Wissenschaften, Moskau (Zeitschrift „Annals of Clinical and Experimental Neurology“ Nr. 3, 2010) [lesen];
Artikel „Neurologische Störungen bei mitochondrialer Enzephalomyopathie – Laktatazidose mit schlaganfallähnlichen Episoden (MELAS-Syndrom)“ von D.A. Kharlamov, A.I. Krapivkin, V.S. Suchorukow, L.A. Kuftina, O.S. Groznova; Moskauer Forschungsinstitut für Pädiatrie und Kinderchirurgie (Zeitschrift „Russisches Bulletin für Perinatologie und Pädiatrie“ Nr. 4(2), 2012) [lesen];
Artikel „Schlaganfallartiger Verlauf der mitochondrialen Enzephalomyopathie (MELAS-Syndrom)“ von I.N. Smirnova, B.A. Kistenev, M.V. Krotenkova, Z.A. Suslina; Wissenschaftliches Zentrum für Neurologie der Russischen Akademie der Medizinischen Wissenschaften, Moskau (Zeitschrift „Nervenkrankheiten“ Nr. 1, 2006) [lesen];
Artikel „Schlaganfälle bei mitochondrialen Erkrankungen“ N.V. Pizova, Abteilung für Nervenkrankheiten mit Kursen in Neurochirurgie und medizinischer Genetik, Staatliche Medizinische Akademie Jaroslawl (Zeitschrift „Neurologie, Neuropsychiatrie, Psychosomatik“ Nr. 9(4), 2017) [lesen];
Artikel „Ischämischer Schlaganfall als Manifestation einer mitochondrialen Enzephalopathie bei einem jungen Patienten“ Murzaliev A.M., Lutsenko I.L., Musabekova T.O., Akbalaeva B.A. (Zeitschrift „Wissenschaft und neue Technologien“ Nr. 6, 2011) [lesen];
Artikel „Epilepsie beim MELAS-Syndrom“ Mukhin K.Yu., Mironov M.B., Nikiforova N.V., Mikhailova S.V., Chadaev V.A., Alikhanov A.A., Ryzhkov B.N., Petrukhin A.S.; Staatliche Bildungseinrichtung für höhere Berufsbildung RSMU Roszdrav; Russisches klinisches Kinderkrankenhaus (Russian Journal of Child Neurology“ Nr. 3, 2009) [lesen];
Artikel „Algorithmus zur Diagnose mitochondrialer Enzephalomyopathien“ von S.N. Illarioshkin, Forschungsinstitut für Neurologie der Russischen Akademie der Medizinischen Wissenschaften (Zeitschrift „Nervous Diseases“ Nr. 3, 2007) [lesen]
© Laesus De Liro
Das MELAS-Syndrom ist eine mitochondriale Erkrankung, die durch eine Schädigung der Muskeln und des Zentralnervensystems gekennzeichnet ist.
MELAS (dt. Mitochondriale Enzephalomyopathie, Laktatazidose und schlaganfallähnliche Episoden – „mitochondriale Enzephalomyopathie, Laktatazidose, schlaganfallähnliche Episoden“) ist eine fortschreitende neurodegenerative Erkrankung, die durch die im Namen aufgeführten Manifestationen gekennzeichnet ist und von polymorphen Symptomen begleitet wird – Schlaganfall , Diabetes, Krampfanfälle, vermindertes Hörvermögen, Herzerkrankungen, Kleinwuchs, Endokrinopathien, Belastungsunverträglichkeit und neuropsychiatrische Störungen.
Geschichte.
Das MELAS-Syndrom wurde erstmals 1984 von Pavlakis und Kollegen beschrieben; Zehn Jahre später veröffentlichten Pavlakis und Mizio Hirano eine Übersicht über 110 Fälle.
Vererbungsart:
mütterlicherseits
Epidemiologie:
Die genaue Häufigkeit der Erkrankung ist nicht bekannt. In der Literatur liegen nur begrenzte Daten zur Inzidenz der Erkrankung vor. In Nordfinnland beträgt die Häufigkeit der A3243G-Mutation 16,3:100.000.
Pathogenese:
Mutationen der mitochondrialen DNA, die die Atmungskette der Mitochondrien steuern, gehen mit einer Störung oxidativer Phosphorylierungsprozesse einher, der wichtigsten Energiequelle für Stoffwechselprozesse in der Zelle.
Klinische Manifestationen
Unter 40 Jahren werden Patienten mit MELAS mit einem vorübergehenden ischämischen Anfall sowie Epilepsie, wiederholtem Erbrechen, Kopfschmerzen und Muskelschwäche aufgenommen. Bei diesen Patienten wird klinisch häufig eine Demenz diagnostiziert.
Das junge Alter und das Fehlen der für einen Schlaganfall charakteristischen Risikofaktoren helfen, über MELAS nachzudenken.
Labordaten
Unter Laktatazidose versteht man einen Anstieg des Laktat- und Pyruvatspiegels.
Visualisierungsdaten
Die Veränderungen im Gehirn ähneln denen eines Schlaganfalls.
Unterschiede zum Schlaganfall
1) Die betroffenen Gebiete stimmen nicht mit den Grenzen der arteriellen Gefäßterritorien überein.
2) Bei wiederholten Anfällen werden die Läsionen an einer anderen Stelle sichtbar.
+ klinische Daten (junges Alter, Fehlen von Risikofaktoren für einen Schlaganfall).
CT
Mehrere hypodense Bereiche, die nicht dem Gefäßgebiet entsprechen.
Verkalkung der Basalganglien (am häufigsten bei älteren Patienten).
Die Atrophie tritt vor dem Hintergrund einer Regression und einer klinischen Verbesserung auf.
MRT
Akuter Myokardinfarkt
Zur Abgrenzung zum Schlaganfall werden ADC und DWI eingesetzt (bei Schlaganfällen ist die Diffusion eingeschränkt (zytotoxisches Ödem) und bei MELAS ist die Diffusion leicht oder unverändert eingeschränkt (vasogenes Ödem).
Beteiligung der subkortikalen weißen Substanz des Gehirns am pathologischen Prozess.
Verschlechterung der Visualisierung der Klarheit der Gyrikonturen und Zunahme des Signals von ihnen auf T2-gewichteten Bildern.
Chronischer Herzinfarkt
Änderungen können symmetrisch oder asymmetrisch sein.
Die fokale Atrophie tritt vor dem Hintergrund einer Regression und klinischen Verbesserung auf.
Am häufigsten sind die Parietal-, Okzipital- und Temporallappen des Gehirns betroffen.
MR-Spektroskopie
Erhöhter Laktatspiegel.



Das MELAS-Syndrom (mitochondriale Enzephalomyopathie, Laktatazidose und Schlaganfall) – mitochondriale Enzephalomyopathie, Laktatazidose und Schlaganfall – wurde erst vor relativ kurzer Zeit (1984) isoliert. Die Krankheit ist mit einer Punktmutation in der mitochondrialen DNA verbunden, die zu 90 % im Gen lokalisiert ist, das für die Synthese von Leucin-Transfer-RNA kodiert, was deren Aufnahme in Proteine der Atmungskette verhindert. Wie bei allen mitochondrialen Erkrankungen ist die Diagnose des MELAS-Syndroms aufgrund der erheblichen Variabilität im klinischen Erscheinungsbild schwierig.
Die wichtigsten klinischen Manifestationen des MELAS-Syndroms sind: Belastungsunverträglichkeit; wiederkehrende schlaganfallähnliche Zustände. Der Begriff „schlaganfallartig“ ist wahrscheinlich auf die Tatsache zurückzuführen, dass in den meisten der beschriebenen Fälle die führende klinische Manifestation Kopfschmerzen mit Erbrechen, Krämpfen, oft mit Bewusstseinsstörungen, die mehrere Stunden bis mehrere Tage anhalten, sind.
Während dieser Anfälle entwickeln einige Patienten häufig neurologische Störungen in Form von Hemianopsie, Hemiparese und selten in Form von Aphasie. Bei der Durchführung einer CT-Untersuchung zeigen 60 % dieser Patienten Herde geringer Dichte und polymorphe Anfälle; biochemische Untersuchung zeigt Laktatazidose; Ausbruch der Krankheit - im Alter von 5-6 Jahren; Der Krankheitsverlauf ist fortschreitend.
Wir präsentieren unsere eigene Beobachtung des MELAS-Syndroms.
Patient A., 6 Jahre alt, wurde mit Beschwerden über eine akut entwickelte Schwäche der rechten Extremitäten, die über mehrere Stunden anhielt, und Kopfschmerzen in die Klinik eingeliefert. Geboren aus der ersten Schwangerschaft. Schwangerschaft und Geburt verliefen ohne Komplikationen. Die frühe psychomotorische Entwicklung des Kindes war altersgerecht. Im Alter von 1 bis 2 Jahren wurden affektiv-respiratorische Anfälle festgestellt. Ab dem 3. Lebensjahr kommt es häufig zu acetonämischen Erkrankungen, die den Höhepunkt der Kopfschmerzen erreichen.
Bei der Untersuchung in der Klinik: motorische Enthemmung, Unruhe, Müdigkeit unter Stress (sowohl geistig als auch körperlich). Neurologischer Status: kraniale Innervation ohne Merkmale, Muskelhypotonie, Asymmetrie der Sehnenreflexe (rechts deutlicher). Es gibt keine Paresen. Es liegt keine Ataxie vor.
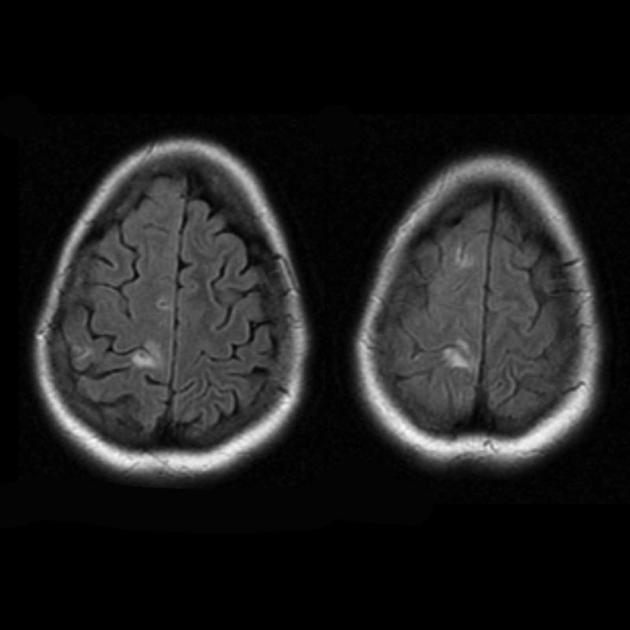
Während der anfänglichen MRT des Gehirns wurde im linken Hinterhauptslappen eine große Zone mit hyperintensivem Signal im T2-Modus und hypointensem Signal im T1-Modus mit klaren Konturen festgestellt; die Mittellinienstrukturen waren nicht verschoben. Dieses radiologische Bild wurde als Phänomen eines ischämischen Schlaganfalls angesehen.
Im Laufe des nächsten Jahres erlitt das Kind dreimal partielle epileptische Anfälle und wiederholte Episoden von paroxysmalen Kopfschmerzen mit Erbrechen. Erhielt eine antikonvulsive Therapie. Ein Jahr nach der ersten Behandlung in der Klinik wurde der Junge aufgrund eines Anfalls generalisierter tonisch-klonischer Anfälle, die zweimal innerhalb einer Stunde auftraten, erneut aufgenommen.
Lethargie und Schwäche nahmen zu und es traten paroxysmale Kopfschmerzen mit einmaligem Erbrechen auf. Die MRT des Gehirns wurde wiederholt – im T1- und T2-Modus wurden Bereiche mit verändertem Signal in den parietookzipitalen Regionen auf beiden Seiten sichtbar gemacht (und die Läsion auf der linken Seite war im Vergleich zu der in der vorherigen MRT-Studie kleiner). So erlitt der Junge in den Monaten nach dem ersten Schlaganfall mindestens drei weitere akute zerebrovaskuläre Unfälle.
Stoffwechselstörungen bestanden in einem deutlichen Anstieg des Laktatspiegels im Blut auf 4,2 mmol/l (normal bis 1,7 mmol/l) und Pyruvat.
Eine umfassende Analyse der Untersuchungs- und klinischen Ergebnisse ermöglichte die Feststellung einer spezifischen nosologischen Form der mitochondrialen Enzephalomyopathie – des MELAS-Syndroms, die erstmals in der Klinik festgestellt wurde.
Anschließend erhielt das Kind eine antikonvulsive Therapie (Topamax 5 mg/kg/Tag), eine Behandlung zur Stimulierung der Gewebeatmung mit Coenzym Q-10, Bernsteinsäure, Cytomac sowie Actovegin und Contrical.
Der vorliegende Fall von wiederkehrenden metabolischen Hirninfarkten bei einem Kind aufgrund des MELAS-Syndroms ist nicht der einzige Fall in der Klinik, und es sammelt sich eine Datenbank ähnlicher Patienten an.
Die Materialien richten sich an Neurologen, Therapeuten und Allgemeinmediziner.
Sergey Likhachev, Leiter, Doktor der Medizin. Naturwissenschaften, Professor;
Inessa Pleshko, führende Forscherin, Kandidatin der medizinischen Wissenschaften. Naturwissenschaften, neurologische Abteilung des Republikanischen Wissenschaftlichen und Praktischen Zentrums für Neurologie und Neurochirurgie.
Die zerebrale autosomal-dominante Arteriopathie mit subkortikalen Infarkten und Leukenzephalopathie (CADASIL) ist eine fortschreitende autosomal-dominante Erkrankung, deren klinische Manifestationen wiederkehrende subkortikale ischämische Schlaganfälle, Migräne, subkortikale Demenz und affektive Störungen umfassen. Aktuelle Prävalenz: 1 Fall
pro 100.000 Einwohner.
Das Republikanische Wissenschaftliche und Praktische Zentrum für Neurologie und Neurochirurgie beobachtet 7 Patienten (darunter 4 Frauen) mit CADASIL; Alter - von 32 bis 68 Jahren. Sie wurden mit neurologischen und molekulargenetischen Methoden untersucht. Es gab charakteristische Symptome; Vorgeschichte von Migräne, wiederkehrenden lakunaren Schlaganfällen und affektiven Störungen. Die MRT des Gehirns zeigte subkortikale Infarkte und eine für CADASIL charakteristische Leukoenzephalopathie.
Bei 2 Personen ergab die molekulargenetische Diagnostik eine heterozygote Mutation im Notch3-Gen auf Chromosom 19, die CADASIL verursacht. Notch-Gene kodieren Transmembranrezeptoren, die an der Zellontogenese beteiligt sind. Bei CADASIL werden in den meisten Fällen Missense-Mutationen festgestellt, wodurch sich die Struktur des Transmembranproteins verändert und seine Funktionen gestört werden.
Die Pathogenese von CADASIL ist nicht vollständig geklärt. Es wird angenommen, dass der Hauptfaktor eine Arteriopathie mit fortschreitendem Verschluss kleiner perforierender Gefäße der weißen Substanz des Gehirns ist (was zu einer chronischen Minderdurchblutung führt). In diesem Fall werden charakteristische körnige osmiophile Einschlüsse gefunden, die zu einer Proliferation der Bestandteile der Basalmembran, einer Verdickung der Tunica media und einer mechanischen Kompression kleiner Arterien führen. Dadurch wird die Blut-Hirn-Schranke geschädigt und es entstehen Ödeme.
Ein weiterer pathologischer Faktor ist die Aktivierung von Astrozyten in der Nähe der Gefäßwand. Sie setzen Endothel-1 frei, was zu einer Gefäßverengung und einer beeinträchtigten Durchblutung führt.
Die Zusammensetzung körniger osmiophiler Einschlüsse ist unbekannt. Es wird angenommen, dass das Notch3-Protein einer ihrer Bestandteile ist. In Hautbiopsien von Patienten mit einer Notch3-Mutation können osmiophile Granula und eine Degeneration glatter Muskelzellen vor dem 20. Lebensjahr nachgewiesen werden.
Klinische Diagnose von CADASIL:
- Familiengeschichte;
- Entwicklung der ersten Krankheitssymptome vor dem 50. Lebensjahr;
- das Vorhandensein von zwei der folgenden Symptome: Migräne, wiederkehrende Schlaganfälle, Stimmungsstörungen, subkortikale Demenz.
Gefäßrisikofaktoren, die ätiologisch mit neurologischen Symptomen assoziiert sind, sollten ausgeschlossen werden. Die MRT zeigt Läsionen der weißen Substanz der Großhirnhemisphären und das Fehlen kortikaler Infarkte.
Eine sichere Diagnose von CADASIL wird durch ein positives Ergebnis der molekulargenetischen Diagnostik oder den Nachweis einer Arteriopathie mit charakteristischen körnigen osmiophilen Einschlüssen im Rahmen einer Haut- oder Muskelbiopsie bestätigt.
Die häufigsten Symptome von CADASIL sind vorübergehende ischämische Anfälle und ischämische Schlaganfälle, die bei fast 85 % der Patienten beobachtet werden.
Sie zeichnen sich durch einen rezidivierenden Verlauf aus, der sich als klassisches lakunares Schlaganfallsyndrom manifestiert und nach einigen Tagen oder Wochen eine vollständige klinische Remission aufweist.
Am zweithäufigsten sind kognitive Beeinträchtigungen (bei 60 % der Patienten festgestellt). Sie können im Alter von 35 Jahren beginnen, manchmal sogar vor ischämischen Episoden. Ungefähr 75 % der Fälle mit CADASIL entwickeln Demenz. Das erste Symptom ist meist eine Migräne; tritt häufig vor dem 20. Lebensjahr auf und geht in der Regel einem Schlaganfall voraus.
Daten zur Beteiligung des Herzens am pathologischen Prozess bei CADASIL sind widersprüchlich. L. Oberstein et al. (2003) fanden heraus, dass 25 % der mit CADASIL diagnostizierten Patienten einen akuten Myokardinfarkt oder eine Q-Wellen-Anomalie im Elektrokardiogramm hatten. In einer anderen Studie haben Cumurciuc et al. (2006) fanden bei 23 Personen mit einer Mutation im Notch3-Gen keine positive Herzanamnese.
Die klinischen Manifestationen von CADASIL und zerebraler Mikroangiopathie anderer Ätiologien sind ähnlich – eine Differentialdiagnose ist erforderlich.
Um CADASIL bei Patienten und ihren Familienangehörigen rechtzeitig zu identifizieren, ist der Rückgriff auf molekulargenetische Methoden und/oder histologische Untersuchungen erforderlich.
MELAS-Syndrom
Die mitochondriale Enzephalomyopathie mit Laktatazidose und schlaganfallähnlichen Episoden (MELAS) ist eine seltene Erbkrankheit, die durch eine Pathologie des mitochondrialen Genoms, eine Störung des Energiestoffwechsels und der Funktion der am stärksten energieabhängigen Organe und Gewebe (ZNS, Herz- und Skelettmuskulatur, Augen, Nieren, Leber, Knochenmark, endokrines System). Die große Variabilität der klinischen Manifestationen des MELAS-Syndroms und sein seltenes Auftreten stellen für einen praktizierenden Arzt Schwierigkeiten bei der Diagnose dar.
Das Republikanische Wissenschafts- und Praxiszentrum für Neurologie und Neurochirurgie beobachtet drei Patienten (eine 46-jährige Frau und ihre Söhne im Alter von 24 und 23 Jahren), bei denen das MELAS-Syndrom diagnostiziert wurde. Sie unterzogen sich einer klinisch-neurologischen Untersuchung, einer molekulargenetischen Diagnostik und einer MRT des Gehirns.
Jeder ist klein; Vorgeschichte von Symptomen einer mitochondrialen Pathologie: Schallempfindungsschwerhörigkeit, migräneähnliche Kopfschmerzen, schlechte Belastungstoleranz. Der Ausbruch der Krankheit sind generalisierte Krampfanfälle. Bei 2 Patienten traten die ersten Symptome vor dem 20. Lebensjahr auf; Es kam zu nacheinander aufeinanderfolgenden epileptischen Anfällen, Episoden von Sehstörungen mit dem Vorhandensein von Herden in der Neurobildgebung im okzipitalen und temporalen Bereich sowie zu erhöhten Laktatwerten im Blut und in der Liquor cerebrospinalis. Eine Person zeigte einen mäßigen Rückgang der kognitiven Funktion; laut Herzultraschall - hypertrophe Kardiomyopathie; Diabetes mellitus.
Eine molekulargenetische Studie ergab für MELAS typische Multisystemläsionen, große Variabilität und unterschiedliche Schweregrade der klinischen Manifestationen, entsprechend der Anzahl mutierter Kopien von A3243G im tRNALeu(UUR)-Gen.
MELAS zeichnet sich durch eine mütterliche Vererbung aus, das Auftreten sporadischer Fälle, wenn eine De-novo-Mutation auftritt; Ansammlung von mitochondrialer DNA in Zellen – sowohl normaler als auch mutierter Typen – (Heteroplasmie) und zufällige Verteilung während der Teilung zwischen Tochterzellen (mitotische Segregation). Auf genetischer Ebene ist die Ursache des MELAS-Syndroms die heteroplasmatische Umlagerung 3243A>G im tRNALeu(UUR)-Gen (in 80 % der Fälle nachgewiesen).
Die Pathogenese der Krankheit ist noch nicht untersucht. Es gibt zwei Haupttheorien: „mitochondriale Angiopathie“ und „mitochondriale Zytopathie“. Es ist bekannt, dass schlaganfallähnliche Läsionen nicht den Gefäßzonen entsprechen und sich aufgrund gleichzeitiger vasogener Ödeme, die durch anhaltende epileptische Aktivität verursacht werden, auf die umliegenden Gebiete ausbreiten. Es wird angenommen, dass schlaganfallähnliche Episoden durch eine neuronale Übererregbarkeit in einem lokalisierten Bereich des Gehirns verursacht werden. Sie entsteht durch eine mitochondriale Dysfunktion in kapillaren Endothelzellen, Neuronen oder Astrozyten; depolarisiert benachbarte Neuronen, was zur Ausbreitung epileptischer Aktivität führt.
Darüber hinaus hat die Einzelphotonen-Emissions-Computertomographie (SPECT) gezeigt, dass Patienten mit MELAS in den Intervallen zwischen schlaganfallähnlichen Episoden eine Minderdurchblutung des hinteren cingulären Kortex aufweisen, was auf eine Störung der zerebralen Hämodynamik hinweist.
Eine beeinträchtigte oxidative Phosphorylierung und eine Störung der mitochondrialen Atmungskette tragen dazu bei, dass der katabole Stoffwechsel vorherrscht und vom Krebszyklus zur anaeroben Glykose mit Laktatansammlung übergeht. Hohe Konzentrationen letzterer im Zentralnervensystem gehen in der Regel mit Perioden neurologischer Symptome einher.
Die wichtigsten klinischen Anzeichen von MELAS sind schlaganfallartige Episoden, Laktatazidose und das Vorhandensein „rissiger roter Fasern“ in Muskelbiopsien. Weitere Manifestationen können Demenz, Psychose, epileptische Anfälle, migräneähnliche Kopfschmerzen, Ataxie, Myopathie, Verkalkung der Basalganglien gemäß Bildgebung, optische Atrophie, Retinopathie, Taubheit, Diabetes, intestinale Pseudoobstruktion, Kardiomyopathie sein.
Das frühe Erkrankungsalter von MELAS liegt zwischen 5 und 20 Jahren, es gibt jedoch Beobachtungen eines späten Auftretens – im 5.–6. Lebensjahrzehnt. Es gibt Fälle, in denen das Syndrom nach Herzerkrankungen begann.
Der Multisystemcharakter der Läsion bei MELAS erschwert die klinische Diagnose.
Der erbliche Charakter der Krankheit erfordert die Durchführung molekulargenetischer Untersuchungen, um eine genaue Diagnose zu stellen.
und identifizieren Sie andere Patienten aus den Angehörigen des Patienten.
Die Materialien richten sich an Neurologen, Therapeuten und Allgemeinmediziner.
 (1 Bewertungen im Durchschnitt: 5,00 von 5)
(1 Bewertungen im Durchschnitt: 5,00 von 5)