Molasses syndrome. Rare diseases. Text of the scientific work on the topic "Epilepsy in melas syndrome"
MELAS syndrome refers to mitochondrial diseases (MD), which are caused by genetic and structural-biochemical defects of mitochondria and are accompanied by impaired tissue respiration and, as a consequence, a systemic defect in energy metabolism, as a result of which the most energy-dependent tissues and target organs are affected in various combinations: brain, skeletal muscles and myocardium, pancreas, organ of vision, kidneys, liver. Clinical disorders in these organs can occur at any age. At the same time, the heterogeneity of symptoms complicates the clinical diagnosis of these diseases. The need to exclude MB arises in the presence of multisystem manifestations that do not fit into the usual pathological process. The frequency of respiratory chain dysfunction is estimated from 1 in 5 - 10 thousand to 4 - 5 per 100 thousand newborns.
read also the post: Mitochondrial diseases(to the website)
MELAS syndrome (Mitochondrial Encephalomyopathy, Lactic Acidosis and Stroke-like episodes) is a multisystem disease characterized by stroke-like episodes occurring at a young age (before 40 years of age), encephalopathy with seizures and dementia, mitochondrial myopathy with the phenomenon of “ragged” red fibers and lactic acidosis (it is possible to increase the level of lactic acid in the blood without acidosis).
MELAS syndrome is caused by point mutations in mitochondrial DNA (mtDNA). The disease is inherited on the maternal side (hence, relatives on the maternal side are likely carriers of such mutations; more often, relatives on the maternal side describe an oligosymptomatic clinical picture with individual symptoms of MELAS syndrome; in asymptomatic relatives, MELAS syndrome is identified only by the results of muscle biopsy or molecular research). Currently, more than ten genes are known whose mutations lead to the development of the clinical picture of MELAS syndrome. In most cases, the development of MELAS syndrome is caused by mutations in genes encoding the functions of transfer RNA.
Typically, the disease debuts between the ages of 6 and 10 years (the age of onset is from 3 to 40 years; early onset of the disease is typical and occurs in 90% of patients). Patients are characterized by short stature (and exercise intolerance). On the part of the internal organs, cardiomyopathy, cardiac conduction disturbances, diabetes mellitus, nephropathy and impaired motility of the gastrointestinal tract may be observed.
Remember! The main clinical criteria for the diagnosis of MELAS are: [ 1 ] maternal type of inheritance; [ 2 ] onset before age 40; [ 3 ] normal psychomotor development before the disease; [ 4 ] exercise intolerance; [ 5 ] migraine-like headache with nausea and vomiting; [ 6 ] stroke-like episodes; [ 7 ] encephalopathy with epileptic seizures and/or dementia (myoclonic seizures are most often recorded, but focal sensory, motor and secondary generalized tonic-clonic seizures are also noted); [ 8 ] lactic acidosis; [ 9 ] ragged red fibers in skeletal muscle biopsies; [ 10 ] progressive course.
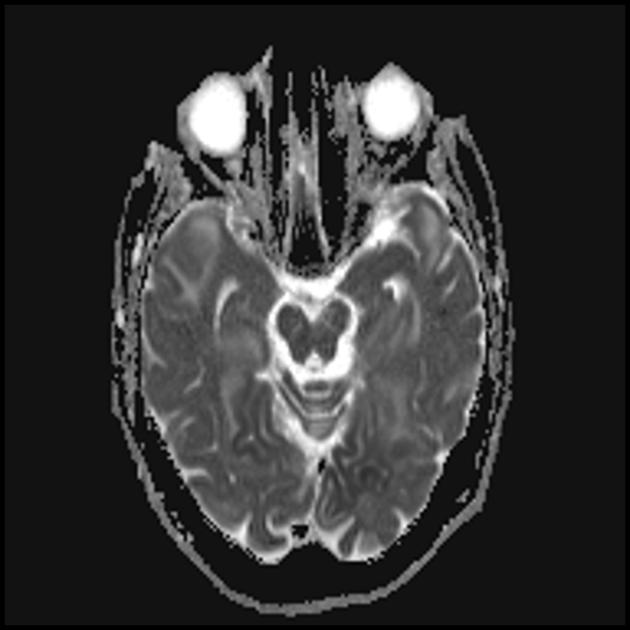
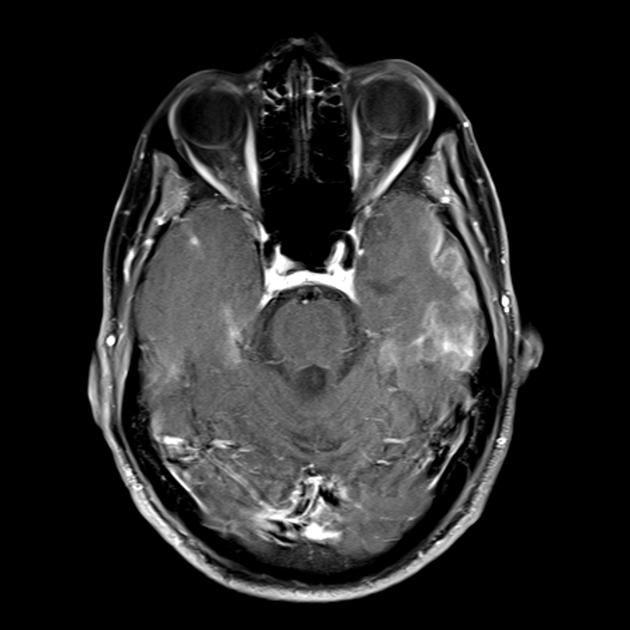
The hallmark clinical feature of MELAS syndrome is stroke-like episodes (IPE), which cause the sudden development of focal neurological disorders. A characteristic feature of IPE is the “posterior” localization of lesions in the brain. Most often, lesions are located in the occipital, parietal and temporal lobes, less often in the frontal lobe, cerebellum or basal ganglia; often they are multiple. The selectivity of the localization of focal changes determines the characteristics of focal neurological symptoms: hemianopsia, sensory aphasia, acalculia, agraphia, optical-spatial disorders, ataxia, changes in consciousness ([ !!! ] most often the lesions are localized in the cortex of the occipital lobes of the cerebral hemispheres, which leads to hemianopsia or cortical blindness). Strokes can resolve or be long-term determined in the form of clinical and/or radiological changes (which depends on the severity of metabolic disorders caused by energy deficiency of neurons). Often repeated “brain infarctions” develop at intervals of 1 to 3 months in symmetrical areas. These lesions can be small or large, single or multiple, usually they are asymmetrical and their localization does not correspond to the area of the blood supply. In addition, patients with MELAS syndrome may have calcifications in the basal ganglia (in these cases, a CT scan of the brain may be helpful). In the neurological status, these morphological changes are manifested by myoclonus, ataxia, episodes of acute psychosis or disturbance (deficit) of consciousness up to coma ([ !!! ] feature of these acute episodes, incl. strokes, on the one hand, a rapid [from several hours to several weeks] regression of symptoms, on the other, a tendency to relapse); from the sensory organs, atrophy of the optic nerves, pigmentary retinopathy and hearing loss are detected.
The following mechanisms are believed to be important in the genesis of IPE: [ 1 ] metabolic disorders in the brain with the development of lactic acidosis due to mitochondrial energy deficiency; [ 2 ] cerebral ischemia caused by mitochondrial angiopathy at the level of small arteries; [ 3 ] local increase in neuronal excitability due to mitochondrial dysfunction in neurons, astrocytes or capillary endothelium, which gradually spreads throughout the cerebral cortex, is combined with the development of edema and can lead to laminar necrosis in the cerebral cortex.
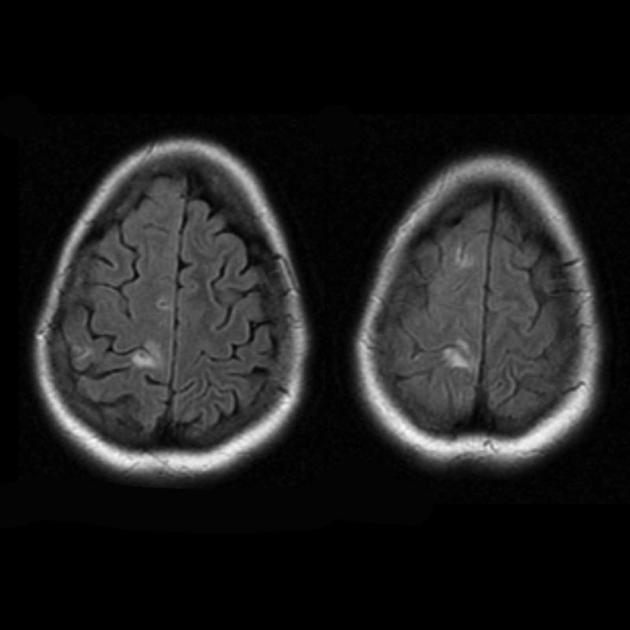
At a superficial glance, a stroke with MELAS syndrome is similar to a normal stroke due to thrombosis or embolism. In fact, stroke-like episodes in MELAS syndrome are atypical: they occur in young people, are often provoked by infectious diseases, and can occur in the form of migraine-like headaches or seizures. MRI scanning of acute IPE in MELAS syndrome reveals abnormalities such as increased signal on T2-weighted or FLAIR (water-attenuated inversion recovery) images. The lesions do not coincide with the territories of the major cerebral arteries, but largely involve the cortex and underlying white matter, with moderate involvement of the deep white matter. Acute brain lesions on MRI in MELAS syndrome may change, migrate, or even disappear ([ !!! ] characterized by fluctuation of foci, determined by MRI). Angiography reveals the absence of significant vascular pathology: in addition to normal results, an increase in the caliber of arteries, veins or capillary hyperemia can be detected.
Neuromorphological studies of the brain in MELAS syndrome show the presence of multifocal necrosis, located mainly in the cerebral cortex and subcortical white matter, as well as in the cerebellum, thalamus and basal ganglia. The lesions resemble areas of infarction, but, as mentioned above, do not coincide with the basins of large cerebral vessels. There is also spongiform degeneration in the cerebral cortex, proliferation of capillaries and depletion of neurons.
Remember! Stroke-like episodes in MELAS have the following features: [ 1 ] young age (usually up to 40 years); [ 2 ] frequent presence of a provoking factor (occurs after febrile fever, epileptic attack, migraine-like headache); [ 3 ] favorite localization - occipital region; [ 4 ] lesions, as a rule, are located outside the area of large cerebral arteries, most often located in the cortex or deep structures of the white matter of the brain.
When making a differential diagnosis of IPE and cerebral infarction, the following symptoms are taken into account:
■ gradual, over several days, increase in focal neurological symptoms (the pathophysiological basis for this pace of development is the gradual increase in energy deficiency of the brain due to impaired oxidative phosphorylation in mitochondria);
■ a gradual decrease in the level of wakefulness, which is in dissonance with a relatively mild focal neurological deficit, is not accompanied by a secondary brainstem syndrome and, therefore, cannot be explained by an increase in cerebral infarction and edema (these symptoms are also based on a metabolic disorder caused by a violation of the energy supply of the brain );
■ development in the acute period of repeated local and generalized epileptic seizures, which, according to the literature, occur in 2/3 of patients with IPE (seizures are not associated with cerebrovascular accident, since the source of their generation is discharge activity in both hemispheres of the brain, and not in structures confined to a specific basin of cerebral arteries; the recurrent nature of seizures and the absence of pronounced persistent focal neurological symptoms are also not characteristic of acute cerebral infarction);
■ complete patency of the cerebral arteries according to duplex scanning of the brachiocephalic arteries and cerebral angiography, which is not typical for ischemic stroke;
■ features of the neuroimaging picture: predominantly cortical localization of foci and their “posterior location”, which is characteristic of MELAS and is explained by the greater vulnerability of neurons in these areas due to their greater energy need; Another neuroimaging feature is the disappearance of some foci, which, apparently, is based on edema, and not necrosis of the brain substance due to metabolic disorders.
![]()
MELAS syndrome on RADIOPAEDIA.org
One of the main manifestations of MELAS syndrome is also muscle weakness (myopathic syndrome). However, the nonspecificity of this symptom does not allow a diagnosis. Only when migraine, seizures and/or stroke-like events occur can the onset of MELAS syndrome be diagnosed.
Screening tests for MELAS syndrome are neuroimaging and a study of the level [increase] of lactate in the blood (partially in the cerebrospinal fluid) - a blood test for lactic (lactate) and pyruvic acid (blood lactate level [normal] - venous blood - 0.5 - 2 .2 mmol/l, arterial blood - 0.5 - 1.6 mmol/l; lactate/pyruvate ratio - 10/1). The diagnosis can be confirmed by DNA testing to determine the most common mutations. In the absence of common point mutations in MELAS syndrome, a muscle biopsy (using Gomori's three-color method to detect ragged red fibers [RRFs] - myofibrils with a high content of the mutant genome and a large number of proliferating altered mitochondria) can help in the diagnosis. It (biopsy) also allows one to determine the presence of biochemical defects in the respiratory chain, mainly associated with the enzymes succinate dehydrogenase and cytochrome oxidase.

Treatment of MELAS syndrome includes two main areas. The first is syndromic therapy (the focus is on epilepsy, diabetes, etc.). It does not differ from generally accepted approaches to the treatment of syndromes. Stopping epileptic seizures is necessary because the metabolic stress that occurs during seizures can trigger the development of stroke-like episodes. Valproic acid derivatives, widely used in epileptology, inhibit mitochondrial functions and their use is undesirable. If it is impossible to discontinue the drug, you should simultaneously take levocarnitine at a dose of up to 100 mg/kg per day. Phenytoin and barbiturates should also be avoided. The second direction of treatment is pathogenetic, but currently there is no effective pathogenetic therapy. The treatment strategy is aimed at improving the energy metabolism of the cell and includes the administration of coenzyme Q or idebinone (Noben), succinic acid preparations, vitamins K1 and K3, nicotinamide, riboflavin, L-carnitine, antioxidants (mexidol, mildronate, vitamins E and C), lactate correctors -acidosis (dimephosphone). [ !!!
] It is necessary to avoid the use of drugs that inhibit mitochondrial function (barbiturates, valproates, statins, glucocorticoids).
Read more about MELAS syndrome in the following sources:
presentation “MELAS syndrome” Kuzenkova L.M., Globa O.V.; Department of Psychoneurology, Research Institute of Pediatrics, Scientific Center for Children's Health, Russian Academy of Medical Sciences, Moscow [read];
article “Mitochondrial encephalopathy with stroke-like episodes and lactic acidosis (MELAS syndrome): diagnostic criteria, features of epileptic seizures and approaches to treatment using the example of a clinical case” Yamin M.A., Chernikova I.V., Araslanova L.V., Shevkun P.A.; State Autonomous Institution of the Rostov Region “Regional Consultative and Diagnostic Center”; Department of Neurology and Neurosurgery with courses of manual therapy and reflexology of the Faculty of Education and Training of the Federal State Budgetary Educational Institution of Higher Education "Rostov State Medical University" of the Ministry of Health of Russia (magazine "Neurology, neuropsychiatry, psychosomatics" No. 9(4), 2017) [read];
article “Mitochondrial cytopathies: MELAS and MIDD syndromes. One genetic defect - different clinical phenotypes” Muranova A.V., Strokov I.A.; Federal State Budgetary Educational Institution of Higher Education “First Moscow State Medical University named after. THEM. Sechenov" Ministry of Health of the Russian Federation, Moscow (Neurological journal, No. 1, 2017) [read];
article “Stroke-like episodes in mitochondrial encephalomyopathy with lactic acidosis” L.A. Kalashnikova, L.A. Dobrynina, A.V. Sakharova, R.P. Tchaikovskaya, M.F. Mir-Kasimov, R.N. Konovalov, A.A. Shabalina, M.V. Kostyreva, V.V. Gnezditsky, S.V. Protsky; Scientific Center of Neurology of the Russian Academy of Medical Sciences, Moscow (journal “Annals of Clinical and Experimental Neurology” No. 3, 2010) [read];
article “Neurological disorders in mitochondrial encephalomyopathy - lactic acidosis with stroke-like episodes (MELAS syndrome)” by D.A. Kharlamov, A.I. Krapivkin, V.S. Sukhorukov, L.A. Kuftina, O.S. Groznova; Moscow Research Institute of Pediatrics and Pediatric Surgery (magazine “Russian Bulletin of Perinatology and Pediatrics” No. 4(2), 2012) [read];
article “Stroke-like course of mitochondrial encephalomyopathy (MELAS syndrome)” by I.N. Smirnova, B.A. Kistenev, M.V. Krotenkova, Z.A. Suslina; Scientific Center of Neurology of the Russian Academy of Medical Sciences, Moscow (magazine “Nervous Diseases” No. 1, 2006) [read];
article “Strokes in mitochondrial diseases” N.V. Pizova, Department of Nervous Diseases with courses in neurosurgery and medical genetics, Yaroslavl State Medical Academy (magazine “Neurology, neuropsychiatry, psychosomatics” No. 9(4), 2017) [read];
article “Ischemic stroke as a manifestation of mitochondrial encephalopathy in a young patient” Murzaliev A.M., Lutsenko I.L., Musabekova T.O., Akbalaeva B.A. (magazine “Science and New Technologies” No. 6, 2011) [read];
article “Epilepsy in MELAS syndrome” Mukhin K.Yu., Mironov M.B., Nikiforova N.V., Mikhailova S.V., Chadaev V.A., Alikhanov A.A., Ryzhkov B.N., Petrukhin A.S.; State Educational Institution of Higher Professional Education RSMU Roszdrav; Russian Children's Clinical Hospital (Russian Journal of Child Neurology" No. 3, 2009) [read];
article “Algorithm for diagnosing mitochondrial encephalomyopathies” by S.N. Illarioshkin, Research Institute of Neurology of the Russian Academy of Medical Sciences (magazine “Nervous Diseases” No. 3, 2007) [read]
© Laesus De Liro
MELAS syndrome is a mitochondrial disease characterized by muscle and central nervous system damage.
MELAS (eng. Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes - “mitochondrial encephalomyopathy, lactic acidosis, stroke-like episodes”) is a progressive neurodegenerative disease characterized by the manifestations listed in the name and is accompanied by polymorphic symptoms - stroke, diabetes, seizures, decreased hearing, heart disease, short stature, endocrinopathies, exercise intolerance and neuropsychiatric disorders.
Story.
MELAS syndrome was first described in 1984 by Pavlakis and colleagues; ten years later, Pavlakis and Mizio Hirano published a review of 110 cases.
Inheritance type:
maternal
Epidemiology:
The exact incidence of the disease is not known. There is limited data on the incidence of the disease in the literature. In northern Finland, the frequency of the A3243G mutation is 16.3:100,000.
Pathogenesis:
Mutations of mitochondrial DNA, which control the respiratory chain of mitochondria, are accompanied by disruption of oxidative phosphorylation processes, the most important source of energy for metabolic processes in the cell.
Clinical manifestations
Under the age of 40, patients with MELAS are admitted with a transient ischemic attack, as well as epilepsy, repeated vomiting, headache, and muscle weakness. These patients are often clinically diagnosed with dementia.
Young age and the absence of risk factors characteristic of stroke helps to think about MELAS.
Laboratory data
Lactate acidosis is an increase in lactate and pyruvate levels.
Visualization data
The changes in the brain are similar to those caused by a stroke.
Differences from stroke
1) the affected areas do not coincide with the boundaries of the arterial vascular territories.
2) with repeated attacks, the lesions are visualized in a different location.
+ clinical data (young age, absence of risk factors for stroke).
CT
Multiple hypodense areas that do not correspond to the vascular territory.
Calcification of the basal ganglia (most common in older patients).
Atrophy occurs against a background of regression and clinical improvement.
MRI
Acute myocardial infarction
To differentiate from stroke, ADC and DWI are used (with strokes, diffusion is limited (cytotoxic edema), and with MELAS, diffusion is limited slightly or without changes (vasogenic edema).
Involvement of the subcortical white matter of the brain in the pathological process.
Deterioration in the visualization of the clarity of the contours of the gyri and an increase in the signal from them on T2-weighted images.
Chronic heart attack
Changes can be symmetrical or asymmetrical.
Focal atrophy occurs against a background of regression and clinical improvement.
The parietal, occipital and temporal lobes of the brain are most often affected.
MR spectroscopy
Increased lactate levels.



MELAS syndrome (mitochondrial encephalomyopathy, lactic acidosis and stroke) - mitochondrial encephalomyopathy, lactic acidosis and stroke - was isolated relatively recently (in 1984). The disease is associated with a point mutation in mitochondrial DNA, which is 90% localized in the gene encoding the synthesis of leucine transfer RNA, which prevents its inclusion in proteins of the respiratory chain. As with all mitochondrial diseases, the diagnosis of MELAS syndrome is difficult due to the significant variability in clinical presentation.
The main clinical manifestations of MELAS syndrome are: exercise intolerance; recurrent stroke-like conditions. The term “stroke-like” is probably due to the fact that in most of the cases described, the leading clinical manifestation is headache with vomiting, convulsions, often with impaired consciousness, lasting from several hours to several days.
During these attacks, a number of patients often develop neurological disorders in the form of hemianopsia, hemiparesis, and rarely in the form of aphasia. When performing a CT scan, 60% of such patients reveal foci of low density and polymorphic seizures; biochemical examination reveals lactic acidosis; onset of the disease - at 5-6 years of age; the course of the disease is progressive.
We present our own observation of MELAS syndrome.
Patient A., 6 years old, was admitted to the clinic with complaints of acutely developed weakness in the right extremities, which lasted for several hours, and headaches. Born from the first pregnancy. Pregnancy and childbirth proceeded without complications. The child's early psychomotor development was age appropriate. From 1 year to 2 years, affective-respiratory paroxysms were noted. From the age of 3 years, frequent acetonemic conditions develop at the height of headache.
During the examination at the clinic: motor disinhibition, restlessness, fatigue under stress (both mental and physical). Neurological status: cranial innervation without features, muscle hypotonia, asymmetry of tendon reflexes (more vivid on the right). There are no paresis. There is no ataxia.
During the initial MRI of the brain, a large zone of hyperintense signal in the T2 mode and hypointensity in the T1 mode with clear contours was detected in the left occipital lobe; the midline structures were not displaced. This radiological picture was regarded as a phenomenon of ischemic stroke.
Over the next year, the child had partial epileptic seizures three times, and repeated episodes of paroxysmal headaches with vomiting. Received anticonvulsant therapy. A year after the first treatment at the clinic, the boy was admitted again due to an attack of generalized tonic-clonic seizures that occurred twice within an hour.
Lethargy and weakness increased, and a paroxysmal headache with single vomiting appeared. MRI of the brain was repeated - in T1 and T2 modes, areas of altered signal were visualized in the parieto-occipital regions on both sides (and the lesion on the left was smaller in size compared to that in the previous MRI study). Thus, in the several months following the first stroke, the boy suffered at least three more acute cerebrovascular accidents.
Metabolic disorders consisted of a significant increase in the level of lactate in the blood to 4.2 mmol/l (normal up to 1.7 mmol/l) and pyruvate.
A comprehensive analysis of the examination and clinical results made it possible to establish a specific nosological form of mitochondrial encephalomyopathy - MELAS syndrome, which was established in the clinic for the first time.
Subsequently, the child received anticonvulsant therapy (Topamax 5 mg/kg/day), treatment aimed at stimulating tissue respiration using coenzyme Q-10, succinic acid, Cytomac, as well as Actovegin and Contrical.
The given case of recurrent metabolic cerebral infarctions in a child caused by MELAS syndrome is not the only case in the clinic, and a database of similar patients is accumulating.
The materials are intended for neurologists, therapists, and general practitioners.
Sergey Likhachev, head, doctor of medicine. sciences, professor;
Inessa Pleshko, leading researcher, candidate of medical sciences. Sciences, neurological department of the Republican Scientific and Practical Center of Neurology and Neurosurgery.
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) is a progressive autosomal dominant disease whose clinical manifestations include recurrent subcortical ischemic strokes, migraine, subcortical dementia and affective disorders. Current prevalence - 1 case
per 100,000 population.
The Republican Scientific and Practical Center for Neurology and Neurosurgery is observing 7 patients (including 4 women) with CADASIL; age - from 32 to 68 years. They were examined by neurological and molecular genetic methods. There were characteristic symptoms; history of migraine, recurrent lacunar strokes and affective disorders. MRI of the brain revealed subcortical infarcts and leukoencephalopathy characteristic of CADASIL.
In 2 people, molecular genetic diagnostics revealed a heterozygous mutation in the Notch3 gene on chromosome 19, which causes CADASIL. Notch genes encode transmembrane receptors involved in cell ontogenesis. With CADASIL, in most cases, missense mutations are determined, due to which the structure of the transmembrane protein changes and its functions are disrupted.
The pathogenesis of CADASIL is not completely clear. It is believed that the main factor is arteriopathy with progressive occlusion of small perforating vessels of the white matter of the brain (leading to chronic hypoperfusion). In this case, characteristic granular osmiophilic inclusions are found, causing proliferation of the components of the basement membrane, thickening of the tunica media and mechanical compression of small arteries. As a result, the blood-brain barrier is damaged and edema develops.
An additional pathological factor is the activation of astrocytes near the vascular wall. They release endothelium-1, causing vasoconstriction and impaired blood flow.
The composition of granular osmiophilic inclusions is unknown. It is assumed that the Notch3 protein is one of their components. In skin biopsies of patients with a Notch3 mutation, osmiophil granules and smooth muscle cell degeneration can be detected before age 20.
Clinical diagnosis of CADASIL:
- family history;
- development of the first symptoms of the disease before age 50;
- the presence of two of the following symptoms - migraine, recurrent strokes, mood disorders, subcortical dementia.
Vascular risk factors etiologically associated with neurological symptoms should be excluded. MRI shows lesions of the white matter of the cerebral hemispheres and the absence of cortical infarcts.
A reliable diagnosis of CADASIL is confirmed by a positive result of molecular genetic diagnostics or the detection of arteriopathy with characteristic granular osmiophilic inclusions during a skin or muscle biopsy.
The most common symptoms of CADASIL are transient ischemic attacks and ischemic strokes, observed in almost 85% of patients.
They are characterized by a recurrent course, manifesting themselves as classic lacunar stroke syndromes and complete clinical remission after a few days or weeks.
The second most common are cognitive impairments (noted in 60% of patients). They can begin at the age of 35, sometimes even before ischemic episodes. Approximately 75% of cases with CADASIL develop dementia. The first symptom is usually a migraine; often occurs before age 20 and usually precedes strokes.
Data on the involvement of the heart in the pathological process in CADASIL are contradictory. L. Oberstein et al. (2003) found that 25% of patients diagnosed with CADASIL had a history of acute myocardial infarction or Q-wave abnormality on the electrocardiogram. In another study, Cumurciuc et al. (2006) found no positive cardiac history in 23 people with a mutation in the Notch3 gene.
The clinical manifestations of CADASIL and cerebral microangiopathy of other etiologies are similar - differential diagnosis is required.
To promptly identify CADASIL in patients and their family members, it is necessary to resort to molecular genetic methods and/or histological studies.
MELAS syndrome
Mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS) is a rare hereditary disease caused by pathology of the mitochondrial genome, disruption of energy metabolism and the functioning of the most energy-dependent organs and tissues (CNS, cardiac and skeletal muscles, eyes, kidneys, liver, bone marrow, endocrine system). The wide variability of clinical manifestations of MELAS syndrome and its rare occurrence predetermine difficulties in diagnosis for a practicing physician.
The Republican Scientific and Practical Center for Neurology and Neurosurgery is observing 3 patients (a 46-year-old woman and her sons, 24 and 23 years old) diagnosed with MELAS syndrome. They underwent a clinical neurological examination, molecular genetic diagnostics, and MRI of the brain.
Everyone is short; history of symptoms of mitochondrial pathology: sensorineural hearing loss, migraine-like headaches, poor exercise tolerance. The onset of the disease is generalized convulsive seizures. In 2 patients, the first symptoms appeared before age 20; there were epileptic seizures following one after another, episodes of visual impairment with the presence of foci on neuroimaging in the occipital and temporal regions, increased lactate levels in the blood and cerebrospinal fluid. One person showed a moderate decline in cognitive function; according to cardiac ultrasound - hypertrophic cardiomyopathy; diabetes.
A molecular genetic study revealed multisystem lesions typical of MELAS, wide variability and varying degrees of severity of clinical manifestations, corresponding to the number of mutant copies of A3243G in the tRNALeu(UUR) gene.
MELAS is characterized by a maternal type of inheritance, the presence of sporadic cases when a de novo mutation occurs; accumulation in cells - both normal and mutant types - of mitochondrial DNA (heteroplasmy) and random distribution during division between daughter cells (mitotic segregation). At the genetic level, the cause of MELAS syndrome is the heteroplasmic rearrangement 3243A>G in the tRNALeu(UUR) gene (detected in 80% of cases).
The pathogenesis of the disease has not yet been studied. There are 2 main theories - “mitochondrial angiopathy” and “mitochondrial cytopathy”. It is known that stroke-like lesions do not correspond to vascular zones and spread to surrounding areas due to concomitant vasogenic edema caused by prolonged epileptic activity. Stroke-like episodes are thought to be caused by neural hyperexcitability in a localized area of the brain. It arises from mitochondrial dysfunction in capillary endothelial cells, or neurons, or astrocytes; depolarizes adjacent neurons, leading to the spread of epileptic activity.
In addition, in the intervals between stroke-like episodes, single photon emission computed tomography (SPECT) has shown that patients with MELAS have hypoperfusion of the posterior cingulate cortex, indicating a disorder of cerebral hemodynamics.
Impaired oxidative phosphorylation and disruption of the mitochondrial respiratory chain contribute to the predominance of catabolic metabolism and changes from the Krebs cycle to anaerobic glycosis with lactate accumulation. High levels of the latter in the central nervous system usually correlate with periods of neurological symptoms.
The main clinical signs of MELAS are stroke-like episodes, lactic acidosis, and the presence of “ragged red fibers” in muscle biopsies. Additional manifestations may be dementia, psychosis, epileptic paroxysms, migraine-like headaches, ataxia, myopathy, calcification of the basal ganglia according to neuroimaging, optical atrophy, retinopathy, deafness, diabetes, intestinal pseudo-obstruction, cardiomyopathy.
The early age of onset of MELAS is from 5 to 20 years, but there are observations of a late onset - in the 5th–6th decades of life. There are cases when the syndrome started after cardiac disorders.
The multisystem nature of the lesion in MELAS complicates clinical diagnosis.
The hereditary nature of the disease requires molecular genetic research to be carried out to make an accurate diagnosis.
and identify other patients from among the patient’s relatives.
The materials are intended for neurologists, therapists, and general practitioners.
 (1 ratings, on average: 5,00 out of 5)
(1 ratings, on average: 5,00 out of 5) 

